1、年度检查概况
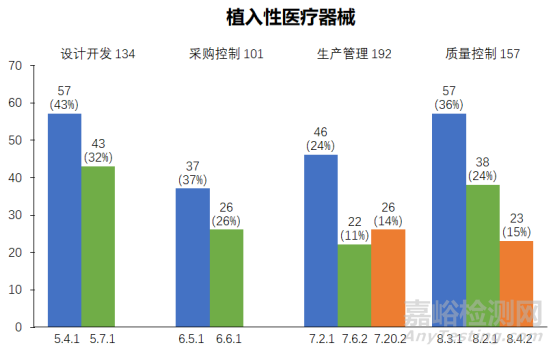
2023年度,上海市器审中心依据《医疗器械生产质量管理规范》和《医疗器械生产质量管理规范附录植入性医疗器械》对植入性医疗器械企业依申请开展现场核查,合计发现不符合项共707项次。从不符合项在《规范》中各章节的分布情况来看,设计开发(134项次)、采购控制(101项次)、生产管理(192项次)、质量控制(157项次)涉及的不符合项占比总数超八成。
2、现场核查常见问题
下面将从设计开发、采购控制、生产管理、质量控制四个方面对植入性医疗器械的常见核查问题进行梳理分析。
2.1设计开发
在设计开发方面,常见问题主要集中在条款*5.4.1和5.7.1。
*5.4.1 设计和开发输出应当满足输入要求,包括采购、生产和服务所需的相关信息、产品技术要求等。
(1)产品图纸、采购质量标准等输出内容不完整。
如产品图纸仅为示意图,无尺寸信息或尺寸信息不完整;当有多种规格时,仅部分规格有图纸。
(2)产品技术要求、产品图纸、采购质量标准、成品检验指导书等技术文件之间存在不一致。
如采购质量标准和图纸规定的材质牌号不一致。
5.7.1 应当对设计和开发进行验证,以确保设计和开发输出满足输入的要求,并保持验证结果和任何必要措施的记录。
(3)设计验证缺项,或发生变更后,未及时进行验证、风险评价。
如产品稳定性验证报告缺少对关键物理、化学等性能的评价或验证,也未提供缺项的依据;未对原验证试验能否覆盖新增规格产品进行评估。
(4)通过与同类产品的验证报告进行等同性评估,但未对产品间差异可能产生的影响进行评价。
如货架有效期通过与已获证产品等同评估,但获证产品材料种类未涵盖本产品的所有材料,企业未对该差异进行评估。
(5)设计开发输出不满足输入的要求。
如某产品技术要求规定静态抗压强度,“试样断裂时最大载荷不低于800N”,但抽查委外验证检测报告中显示断裂最大载荷为700N,输出不满足输入要求。
2.2采购控制
在采购控制方面,常见问题主要集中在条款6.5.1和6.6.1。
6.5.1采购时应明确采购信息,清晰表述采购要求,包括采购物品类别、验收准则、规格型号、规程、图样等内容。
(1)质量协议或采购要求内容不完整,如未明确具体的物料、包装形式、验收要求。
如未明确物料材质、牌号信息,未规定色母要求等;和代理商签订的采购质量协议中,未明确实际生产商信息;初包装材料(如透析纸、盘管等)未明确初始污染菌和微粒要求。
(2)质量协议内容和图纸等技术文件的规定不一致。
如质量标准和图纸对同一原材料规定的材质、牌号不一致。
6.6.1应当对采购物品进行检验或验证,需要进行生物学评价的材料,采购物品应当与经生物学评价的材料相同。
(3)进货检验或验证执行不规范,实际执行和文件规定不一致。
如抽查进货检验记录,检验使用方法和进货检验规程要求的不一致;抽样量与规程要求不一致。
(4)记录不完整。
如未记录进货检使用试剂批号。
2.3生产管理
在生产管理方面,常见问题主要集中在条款*7.2.1、7.6.2和*7.20.2。
*7.2.1应当编制生产工艺规程、作业指导书等,明确关键工序和特殊过程。
(1)作业指导书未明确某些工艺参数。
如内包装封口机有多个参数可调,标准操作程序仅规定封口温度,未规定封口速度、压力。
(2)对关键工序和特殊过程的重要参数缺少验证或确认。
如未对注塑工艺中注射压力、时间、射出速度等参数进行验证;缺少对工艺参数上下限的验证。
7.6.2生产记录应当包括:产品名称、规格型号、原材料批号、生产批号或产品编号、生产日期、数量、主要设备、工艺参数、操作人员等内容。
(3)生产记录信息不完整。
未记录生产设备信息;未记录程序号;未记录工艺参数等。
*7.20.2灭菌过程应当按照相关标准要求在初次实施前进行确认,必要时再确认,并保持灭菌过程确认记录。
(4)未按标准要求进行灭菌确认。
如未对环氧乙烷灭菌过程中关键参数如温度、湿度的上下限开展验证;环氧乙烷灭菌验证记录中未清晰描述装载方式、生物指示剂的放置位置。
2.4质量控制
在质量控制方面,常见问题主要集中在条款*8.3.1、8.2.1和8.4.2。
*8.3.1应当根据强制性标准以及经注册或备案的产品技术要求制定产品的检验规程,并出具相应的检验报告或证书。
(1)检验规程不完整,未明确检测方法、检验条件、接收标准、抽样规则等,不能有效指导检验。
如化学性能检验明确浸提液制备方法;未规定周期检项目检验周期;
(2)检验规程和强制性标准、产品技术要求不一致,或风险评估不充分。
如出厂检验指导书和产品质量控制方式清单规定的抽样方案不一致。
(3)检验规程的执行不规范,实际执行和文件规定不一致。
如未按规定频次实施检测;未按规定数量进行测试;未按规定参数实施检测;未按规定进行检测结果判定。
8.2.1应当定期对检验仪器和设备进行校准或检定,并予以标识。
(4)校准项目范围不完整,校准范围未覆盖实际使用范围。
如万能拉伸试验机校准报告仅校验力值,未对拉伸速率与位移进行校准;
8.4.2检验记录应当包括进货检验、过程检验和成品检验的检验记录、检验报告或证书等。
(5)检验记录信息不完整。
如某检验项目作业指导书要求记录摩擦力平均值和最大值,但检验结果仅记录“合格”,未提供原始测试数据。
3、总结与建议
医疗器械质量体系管理是实现对医疗器械生产全过程控制,保障医疗器械安全有效的重要手段。通过对现场核查常见不符合项的汇总分析,建议企业在日常质量体系运行过程中,应确保各阶段程序受控、分工职责明确、过程完整、内容真实、记录可追溯,落实产品质量主体责任。