原料药杂质谱分析是药物开发中的重要环节,有利于了解药物的安全性和有效性,有助于保证和提高药品的质量。杂质谱分析是杂质研究工作的基础环节,全面的杂质谱分析,可为药品制备工艺的开发和优化、质量控制策略的制定提供指导,可使杂质检查工作有的放矢,是建立合理可行检查方法的基础。笔者结合法规指南要求和自身经验总结出以下五大方面杂质谱分析的研究要点:
一、 杂质的来源及其控制策略
了解杂质的来源,可以帮助研究者减少或规避其产生。杂质可能来自原料、辅料、残留溶剂、生产工艺、包装材料等。因此,对制药过程中的所有环节中杂质的潜在来源之处全面分析,并实施有效地控制是减少或避免杂质产生的关键。根据《中国药典》通则9102,杂质分为有机杂质、无机杂质、残留溶剂、异构体杂质、多晶型杂质和基因毒性杂质,不同类型的杂质来源略有不同,控制策略更是“因杂质而异”。以下是不同类型杂质来源的分析和控制策略简介:
1.1 有机杂质
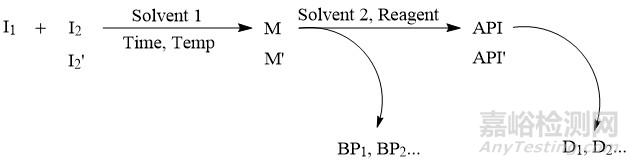
▲图1-化学原料药的合成工艺示意图
原料药中有机杂质的来源往往通过合成工艺路线来综合分析,比如图1是一个化学原料药的合成工艺示意图,根据此反应路线,原料药中有机杂质的潜在来源主要包括以下几个方面:①起始物料(I1、I2)及中间体(M);②起始物料引入的杂质(如I2′),以及起始物料引入杂质的后续反应产物,如I2′的后续反应产物M′、API′;③副产物,如BP1、BP2等等,如果副产物可随主成分一同参与后续的反应,还需关注其后续反应产物;④原料药的降解产物,如D1、D2等等;⑤反应中使用的试剂、配位体、催化剂等等。由于降解产物的化学结构一般与活性成分类似或具有渊源关系,通常也称为有关物质。除了降解产物外,其他4类有机杂质都与制备工艺有关,也称为工艺杂质。
1.1.1 工艺杂质分析
(1)起始物料引入的杂质及后续反应产物
对于外购起始物料,建议根据供应商提供的制备工艺,对可能引入的杂质进行全面的分析和检测,注意分析起始物料引入杂质在后续工艺步骤中的去向/清除情况,结合后续中间体中控数据的积累,合理制定起始物料引入杂质的质控策略(源头控制,过程控制,或在终产品中继续关注)。
除起始物料引入杂质外,还建议重点关注那些可引入后续反应的潜在杂质,通常这类杂质的结构和理化性质与主成分类似,可随主成分一同进行后续的化学反应,后续工艺步骤对其清除难度相对其他杂质而言较高,故在终产品中残留的可能性也较大,这类杂质可与有关物质同时进行检查方法开发与验证。
(2)副产物
建议根据工艺开发过程中掌握的工艺认知、对所涉及化学反应机理的理解以及数据的积累,对各步骤可能产生的副产物进行合理分析,并跟踪其在后续工艺步骤中的去向/清除情况,根据多批次跟踪数据的积累,合理制定各工艺副产物杂质的质控策略。同样,建议重点关注与主成分结构类似、可引入后续反应的副产物杂质。
(3)反应中使用的试剂、配位体、催化剂等物料
当某一种或多种杂质排除为起始物料引入杂质、副产物杂质和降解产物杂质后,需要考虑是否为反应中使用的试剂、配位体、催化剂,或后处理中使用的树脂、吸附剂(如活性炭)等物料引入,建议对每一种物料进行可能性分析、逐一排查,当确定杂质由某种物料引入后,应当评估是否更换甚至禁用该物料,必要时改变工艺,以达到减少或清除该杂质的目的。
1.1.2 有关物质分析
建议可通过结构特征的分析以及试验的手段来研究潜在的降解途径和降解产物,正式稳定性试验、强制降解试验是常用的试验手段。相对于正式稳定性试验,强制降解试验可在较短的时间内获得大量的有益信息。因此在早期研发阶段,强制降解试验是研究潜在降解途径和降解产物的一种有效手段。通过强制降解试验,得出很多潜在的降解杂质和降解途径,可为制定控制这些杂质的策略提供有效的依据,如工艺优化、选择合适的储存条件。
1.2 无机杂质
无机杂质包括催化剂、配位体和试剂;重金属元素杂质(主要产生途径为在药物生产过程中接触到的容器引入或者使用试剂和催化剂等时引入);无机盐;其他物质(例如过滤助剂、活性炭等)。其中重金属元素杂质为无机杂质中最主要的杂质,本部分也将以此种类型杂质为主进行论述。
1.2.1 元素杂质的潜在来源
根据ICH Q3D,在药品生产中,元素杂质的潜在来源广泛,包括:
(1)在原料药生产中有意添加元素(如催化剂)的残留;
(2)非有意添加但在原料药生产所用起始物料、中间体、水或其他试剂中可能存在的元素杂质;
(3)生产设备可能引入到原料药中的元素杂质;
(4)包装系统可能浸出至原料药的元素杂质;
(5)环境中可能引入至原料药中的元素杂质。
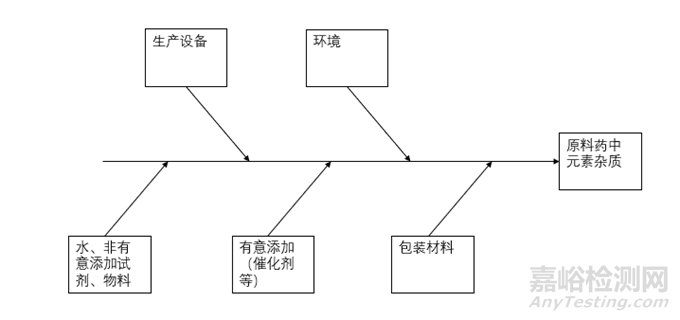
来源途径可以由图2简单概括。通过对上述每一种潜在的来源进行分析,元素杂质可以任何单独或组合的形式被引入到药品中。风险评估过程中,元素杂质的每一种来源的潜在贡献都需考虑,以确定元素杂质对药品的整体贡献情况。
▲图2-原料药中元素杂质潜在来源途径示意图
1.2.2 风险评估
ICH Q3D中表5.1列出了风险评估中建议考虑的元素杂质,共24种。该表适用于药品中所有来源的元素杂质。该表中未列出的元素杂质往往危害性较低,在自然界中存在的含量较高,如钠、钾、钙、镁、磷、硅、硼等,通常不需进行风险评估。
根据潜在元素杂质的识别过程,存在以下两种可能结果:
(1)风险评估过程未识别出任何潜在的元素杂质。应记录风险评估结论和支持性信息及数据;
(2)风险评估过程识别出一个或多个潜在的元素杂质。对于该过程中识别出的任何元素杂质,风险评估均需考虑元素杂质来源多样性,并记录评估结论和支持性信息。
原料药、中间体、起始物料、包装系统和生产设备供应商提供的关于潜在元素杂质的信息有助于申请人的风险评估。支持该风险评估的数据来源包括先验知识、公开发表的文献、相似工艺的数据、供应商信息或数据、原料药、中间体和起始物料的检验数据等。影响原料药中潜在杂质水平的因素也需在风险评估中予以考虑。这些因素包括在后续工艺过程中清除元素杂质的有效性、元素的天然丰度(对于非有意添加类的元素尤为重要)、对于特定来源的元素杂质浓度范围的先验知识等。
1.2.3 控制
元素杂质的控制是药品整体控制策略的一部分,用以确保元素杂质不超过PDE 值(见ICH Q3D中表A.2.1,未列入表中的元素的PDE值可通过ICH Q3D中公式A.1.1计算或查阅相关法规、文献)。当元素杂质水平超过控制阈值(PDE的30 %)时,需采取额外的措施来确保元素杂质水平不超过PDE值。常用的方法包括但不限于:
(1)调整生产工艺步骤,通过特定或非特定的纯化步骤将元素杂质降低至控制阈值之下;
(2)实施工艺过程中或上游控制,旨在将原料药中元素杂质的浓度限制在控制阈值以下;
(3)建立合成中间体的标准限度;
(4)建立起始物料的标准限度。
各个元素杂质的含量应根据 ICH Q6A 的原则进行定期检测,通常每半年一次。
1.3 残留溶剂
1.3.1引入途径
原料药中残留溶剂的可能引入途径主要有以下几种:
(1)合成原料或反应溶剂;
(2)作为反应副产物引入,如甲酯水解生产甲醇;
(3)其他合成原料或其他溶剂带入,如甲苯、苯胺中的少量苯;
(4)其他物质(如大孔吸附树脂中残留的苯、甲苯等)。
1.3.2 分类和限度
根据ICH Q3C,残留溶剂基于风险评估,按照对人体健康的潜在危害,分为三类:
1 类溶剂:应避免的溶剂
此类溶剂为已知的人体致癌物,强疑似人体致癌物,以及环境危害物,共包括苯、四氯化碳、1,2-二氯乙烷、1,1-二氯乙烯、1,1,1-三氯乙烷五种,1类溶剂均有明确的限度,必须严格控制在限度以下。
2 类溶剂:应限制的溶剂
此类溶剂为非遗传毒性动物致癌物,或可能导致其他不可逆毒性如神经毒性或致畸性的溶剂。可能有其他严重但可逆的毒性的溶剂,共包括乙腈、氯苯、氯仿等共31中溶剂。2类溶剂的限度可通过PDE法进行计算,计算方法参照ICH Q3C中3.3章节内容。
3 类溶剂:低潜在毒性的溶剂
此类溶剂为对人体低潜在毒性的溶剂,包括乙酸、丙酮、甲酸、乙醇、乙酸乙酯等应受GMP或其他质量要求限制的常见溶剂(见ICH Q3C中表3)和石油醚、三氯乙酸、三氟乙酸等无足够毒理学数据的溶剂(见ICH Q3C中表4)。前者由于早已被公认为低毒性,对人类健康危害较低,PDE值可认定为≥50 mg/d,然后按照2类溶剂的计算方法得出其限度;后者无PDE值,可通过公式ICH Q3C的附录3中公式(1)计算得出,然后同样按照2类溶剂的计算方法得出其限度。
1.3.3 控制
若某种残留溶剂超出限度,主要的控制措施如下:
(1)优化工艺,减少该溶剂用量,想方设法控制在限度以下;
(2)改变工艺,采用其他的替代溶剂或采用混合溶剂,如1类溶剂超出限度,用2类或3类溶剂替代;2类溶剂超出限度,用限度更宽的其他2类溶剂或3类溶剂替代;3类溶剂超过限度,用其他3类溶剂或水或有机/水混合溶剂替代;
(3)实施工艺过程中或上游控制,旨在将原料药中残留溶剂的浓度限制在限度以下;
(4)制定合成中间体的残留溶剂标准限度;
(5)制定起始物料的残留溶剂标准限度。
1.4 异构体杂质
异构体杂质一般在手性药物中多见,此类杂质的研究是杂质研究中的一个难点。根据《手性药物质量控制研究技术指导原则》报道,异构体杂质的来源主要分为三种途径:直接从起始原料或试剂中引入、不对称合成和消旋体的拆分。在手性药物制备工艺研究中,如果能充分考虑从工艺中对产品的光学纯度进行有效的全程控制,就能从源头上控制产品的质量。尤其是当终产品难以全面有效地控制其光学纯度时,就更应该重视制备工艺研究中的过程控制。
1.5 多晶型杂质
原料药的多晶型主要受不同析晶溶剂、析晶温度、析晶时间、析晶操作条件影响。另外,干燥、粉碎、制粒、球磨等工艺步骤,温度、湿度、光照等环境因素的作用下,可能出现转晶现象,因此多晶型的杂质来源较广泛。控制多晶型杂质的措施主要还是从析晶工艺入手。是否将多晶型检查项订入质量标准可参考《化学仿制药晶型研究技术指导原则(试行)》中决策树1和决策树2。
1.6 基因毒性杂质
基因毒性杂质的来源和控制策略与有机杂质类似,关键和前提是识别某杂质是否为基因毒性杂质,然后采取对应的控制措施。具体识别和控制方法参考ICH M7中表1对此类杂质的分类和控制。
二、 杂质的识别与定性
杂质识别是指通过各种分析方法确定杂质的化学结构,如结构确证。杂质定性则是通过各类谱图(如MS, NMR等)来推断杂质的结构、确定杂质在谱图中的位置和判断原料药中是否含有某种杂质。各类杂质的识别和定性方法如下:
(1)有机杂质和基因毒性杂质的识别通常用MS、NMR、UV、IR四大谱,有时将TGA、DSC、有机元素分析法等作为辅助;定性通常采用LC、GC、MS、UV,其中基因毒性杂质因限度很低(大多ppb级),往往需要很强的灵敏度,故通常采用MS分析;
(2)无机元素杂质的识别和定性通常采用ICP-OES或ICP-MS,其中ICP-MS具有更高的灵敏度、分辨力和专属性;
(3)残留溶剂通常采用GC和GC-MS,对于苯、四氯化碳等限度较低的溶剂,建议选择灵敏度更高的GC-MS;
(4)手性杂质的识别分为直接法和间接法:直接法指只需通过某单一方法即可确证手性药物的构型,主要指单晶X射线衍射法(SXRD);间接法是指仅靠对待测物进行分析,尚难以确证其构型,而需综合其它数据,如与其同系物的相关分析数据相结合才能确定待测物的构型。如化学相关法、比旋度、手性色谱、核磁共振、旋光光谱(ORD)、圆二色谱(CD)等。手性杂质的定性方法主要包括比旋度、使用手性色谱柱的高效液相色谱法(HPLC)或气相色谱法(GC)、化学相关法;
(5)多晶型杂质的识别和定性主要通过单晶X射线衍射法( SXRD)和粉末X射线衍射法(PXRD)。此外,热分析方法(如DSC、TGA和热台显微镜法等) 、光谱法(如IR、Raman和ssNMR等)、 毛细管熔点法(MP)和偏光显微法(PM)均可作为辅助手段进一步支持不同晶型的确证。
三
杂质谱的建立
基于对所有杂质潜在来源的分析、识别和定性后,针对每一种原料药,都应建立一套完整的杂质谱,包括所有可能的杂质,这需要一个大规模的研究工作。有了这个谱,就可以在药品生产过程中快速定性杂质,及时进行控制。
四、 定量分析及限度制定
杂质的存在虽然难以避免,但是必须限制其在药品中的含量。因此,需针对杂质在产品中的最大限度,开发出合适的定量分析方法。
4.1 限度制定
原料药中有机杂质的限度制定通常参考ICH Q3A中附件1的表格,低于报告限的杂质可不进行控制;无机元素杂质的限度制定通常参考ICH Q3D中的第7章内容;残留溶剂的限度制定通常参考ICH Q3C中第4章内容;基因毒性杂质的限度通常参考ICH M7中第7章内容或根据毒理学数据(如CPDB、Toxnet等)来制定;异构体杂质和多晶型杂质由于指导原则中并没有明确的限度制定说明,需要研究者们查询其他的文献资料来制定,如仿制药的限度制定可参考已有的国家标准或国外标准或原研药的质量标准;而对于创新药,就更需要研究者们多下功夫,若权威性的文献资料查不到,可在反复实验基础上,依据经验值来确定杂质限度或者基于药理毒理试验来确定杂质限度。
4.2 定量分析方法
杂质的定量分析方法用于准确、及时地监测药品中杂质的含量,以便必要时采取控制措施。通常定量分析方法与定性分析方法一致,关键是根据杂质限度和重要程度(如存在的可能性、清除难易、是否为特定杂质)选取最准确的定量方式。常用的定量方式包括峰面积归一化法、外标法、主成分自身对照法、内标法、标准加入法等等。
五、 方法验证与稳定性研究
开发出的定量分析方法需要进行验证,以确保方法的可靠性。另外,还需要研究原料药的稳定性,以便了解在药品贮存期间,杂质的变化情况。
方法验证通常参考《中国药典》指导原则9101、USP<1225>、ICH Q2等指南,杂质的主要验证项目有专属性、精密度、准确度、检测限/定量限、耐用性等。另外,元素杂质由于其特殊性,应以USP<233>或EP 2.4.20为主进行方法验证。
方法验证结束后,该方法将一直用于工艺验证和稳定性研究期间杂质的检测。稳定性研究主要参考ICH Q1和《中国药典》指导原则9001进行,根据指南要求,应当首先进行影响因素试验,以便了解影响杂质稳定性的因素及可能的降解途径与降解产物,为生产工艺、贮存条件和是否需要变更降解产物分析方法提供科学依据,也为后续的加速试验和长期试验条件提供参考。而加速试验和长期试验的数据将直接为原料药的贮存、运输条件提供科学依据。
小结
杂质研究是贯穿于药品研发始终的一项重要内容,直接影响药品的有效性和安全性,杂质谱分析是杂质研究工作的基础,基于杂质谱分析的杂质控制是“质量源于设计”基本理念在杂质研究与控制中的一种具体实践。合理的杂质谱分析策略有助于节省研究时间和全面地分析杂质,更有利于药品的成功注册申报。通过本文的讨论,笔者希望给广大同行们一些有益的启示—如何从杂质谱分析入手确立科学的杂质研究思路。