基于全球出现的一系列创新型生物制品注册分类与产品实际用途不匹配的问题,如预防用单克隆抗体和治疗用疫苗,本文对国内外生物制品的分类和注册申请路径进行比较,分析如何与时俱进地调整注册分类方法,为创新生物制品的研发注册提供适宜的监管路径和程序。
《生物制品注册分类及申报资料要求》(2020年第43号)规定,生物制品是指以微生物、细胞、动物或人源组织和体液等为起始原材料,用生物学技术制成,用于预防、治疗和诊断人类疾病的制剂。随着科学技术不断进步,生物制品更新迭代迅速,生物制品治疗用和预防用的功能逐渐交叠,单一类别的生物制品已经无法用治疗或者预防的功能进行固化界定,全球范围出现一系列创新型生物制品,如预防用单克隆抗体(以下简称为单抗)和治疗用疫苗等。随着新型生物制品的出现,无法与现行生物制品注册分类进行匹配。本文对国内外生物制品的分类和注册申请路径进行比较,分析如何与时俱进调整注册分类方法,为创新生物制品的研发注册提供适宜的监管路径和程序。
1、我国生物制品定义和注册分类情况
我国生物制品注册分类大体上分为2个阶段,第1阶段按产品列举方式分类,第2阶段按生物制品预期用途进行分类。
1.1生物制品注册按产品列举式分类
1951年颁布的《生物制品法规》是我国最早的一部关于生物制品的法规,含12个通则和26个品种各论,是最早与疫苗有关的质量标准。1985年之前,我国的生物制药产业仍以传统生物制品为主,即疫苗和血液制品,总体上这个阶段批准的生物制品还是(预防用)疫苗占主导地位。
1985年卫生部颁布了第一部《新生物制品审批办法》,首次将新生物制品按照产品类别划分为4项注册分类。1988年颁布《新生物制品审批办法补充规定》,新生物制品与新药品难以区分者按以下办法处理。原则上按其用途,用作免疫预防者属于生物制品;用作临床治疗者属于药品;如该产品的生产工艺、质量控制标准及其检测方法接近生物制品者,虽属治疗用品仍按新生物制品审批程序申报,申报时需参考《新药审批办法》中有关技术要求。1993年原卫生部颁布《生物制品管理规定》,仍沿用产品列举式分类。
1.2生物制品注册按预期用途分类
随着生物制品种类增多,按产品列举式分类变得愈发困难,1999年新修订的《新生物制品审批办法》中将生物制品申报按照用途分为治疗用新生物制品、预防用新生物制品和体外诊断用品。
2002年,国家药品监督管理局颁布《药品注册管理办法(试行)》(局令第35号) ,细化了生物制品的注册分类,将治疗用生物制品分为15类,包括单抗、基因治疗、体细胞治疗及其制品等。预防用生物制品也分为15类,均为疫苗。2005年,原国家食品药品监督管理局颁布《药品注册管理办法》以及2007年修订均延续了上述分类方式。
《药品注册管理办法》2020年版自2020年7月1日正式实施。生物制品注册按照生物制品创新药、生物制品改良型新药、已上市生物制品(含生物类似药) 等进行分类。为规范生物制品注册申报和管理,《生物制品注册分类及申报资料要求》(2020年6月)将生物制品分为预防用生物制品、治疗用生物制品和按生物制品管理的体外诊断试剂。其中,预防用生物制品是指为预防、控制疾病的发生、流行,用于人体免疫接种的疫苗类生物制品,包括免疫规划疫苗和非免疫规划疫苗,见表1。
2、新型生物制品带来的注册分类问题和挑战
随着生物制药技术的飞速发展,越来越多的新型生物制品申报上市,一方面,以单抗为代表的非疫苗类预防用生物制品出现;另一方面,同一类生物制品可能同时兼有预防用和治疗用等多重功能。目前生物制品的分类方式,将预防用和治疗用功能截然分开,且预防用生物制品等同为疫苗可能导致新型生物制品的注册分类难题。
2.1新型长效单抗无法纳入预防用生物制品类别
单抗是由单一杂交瘤细胞产生,针对单一抗原表位的特异性抗体。自1986年首个鼠源单抗muromonab-CD3经美国FDA批准上市用于治疗器官移植后同种异体排斥反应至今,全球已有100多种单抗获批上市,单抗在癌症、自身免疫性疾病和感染性疾病等疾病中发挥重要作用。
长期以来,单抗不可避免的高研发成本所致的昂贵售价并不亲民,并且频繁地注射给药也给患者带来诸多不便。因此,近年来,延长单抗半衰期成为抗体工程改造的重点。单抗半衰期的延长不仅可以减少给药频率,降低治疗费用,并且使单抗药物用于感染性疾病的预防变为可能。单抗原来半衰期约为11~30d,经抗体工程改造后的单抗半衰期长达2~4个月。目前的生物制品分类方式,预防用生物制品等同为疫苗可能导致全球范围内出现的创新型预防用长效单抗无法纳入预防用生物制品类别。
2.1.1预防呼吸道合胞病毒(respiratory syncytial virus,RSV)感染长效单抗
RSV是全球5岁以下儿童急性下呼吸道感染最常见的病毒病原,也是导致1岁以下婴儿罹患下呼吸道感染住院的首要病毒病原。目前,尚无RSV疫苗和有效的抗病毒药物用于RSV感染的防治。一种可选择的预防RSV的方法是用单抗进行被动免疫预防。帕利珠单抗是目前唯一被FDA批准用于RSV被动免疫预防的单抗,仅用于患有先天性心脏病或肺部疾病的不足35周的早产儿,且半衰期短,需要每周注射。长效、全人源预防用RSV单抗一直是全球各国研发的热点。预防用长效单抗nirsevimab是首个完成全球Ⅲ期临床试验的单抗,与帕利珠单抗相比中和RSV活性高50倍,具有超长半衰期,并扩大适用人群为所有婴幼儿。其中,Ⅲ期临床研究证明单次肌内注射nirsevimab可在整个RSV流行季,给晚期健康早产儿及足月儿提供持续5个月的保护。
目前,我国已有3种用于预防RSV引起的下呼吸道疾病的长效单抗获批进入临床试验阶段,目标人群包括0~2岁的健康婴幼儿、早产儿和具有RSV高危因素的婴幼儿。尽管3款种长效单抗均通过被动免疫用于疾病预防,但由于不是疫苗,不符合我国目前预防用生物制品(等同为疫苗)的注册分类,无法纳入预防用生物制品类别,因此在临床试验受理阶段暂纳入治疗用生物制品注册分类,见表2。

2.1.2预防疟疾感染长效单抗
疟疾是疟原虫感染所致的地方性传染病,主要流行于热带和亚热带地区,疾病病死率较高。根据世界卫生组织最新报告,2019年全球报告2.29亿例疟疾病例,逾40.9万例死于疟疾。目前已有3个用于预防(和治疗)疟疾感染的单抗进入临床试验阶段(见表3),其中长效单抗L9LS是CIS43LS的下一代单抗,其针对的是CSP-1蛋白的一个更紧密的区域,具有高亲和力,保护效力是CIS43LS的3倍。meplazumab是靶向CD147的单抗药物,2020年1月获美国FDA批准开展用于治疗和预防严重疟疾的Ⅰ期临床试验。

2022年8月4号,一项旨在预防疟疾感染的新型单抗L9LS的Ⅰ期临床试验结果在《新英格兰医学杂志》上发表。试验旨在评估L9LS对从未患过疟疾或未接种疟疾疫苗的健康成年人的安全性和药动学,以及人疟疾感染的保护效力。试验通过皮下注射或静脉注射给予L9LS,在接受抗体注射的2~6周内,让所有参与者都暴露于携带疟原虫的蚊子,暴露后几周内,研究人员观察到几乎所有的受试者都得到了保护。L9LS经抗体工程优化(LS突变),半衰期为56d,对于5岁以下的儿童,1次皮下注射可能提供至少6个月的保护效力。
2.1.3新型冠状病毒肺炎(COVID-19)暴露前预防长效单抗
COVID-19暴发以来,疫苗在控制病毒传播和降低死亡率方面发挥着重要作用,是向大多数人提供COVID-19保护的最适当措施。此外,在未接种疫苗或对疫苗有不良反应的人群(如免疫功能低下的个人)中,长效单抗可提供暴露前被动免疫预防作用。
恩适得(Evusheld,tixagevimab与cilgavimab的组合)是目前全球唯一一款用于COVID-19暴露前预防的单抗组合药物,2021年12月获得美国FDA的紧急使用授权,2022年3月获得欧洲EMA上市许可,2022年6月通过海南博鳌乐城国际医疗旅游先行区特殊进口审批,适用于成人和青少年(年龄≥12岁且体重≥40 kg)的新冠病毒暴露前预防。
Evusheld获批美国FDA紧急使用授权是基于PROVENTⅢ期暴露前预防临床试验数据,与安慰剂相比,Evusheld组受试者出现COVID-19症状的风险在统计学上显著降低(初步分析为77%,中位6个月随访时为83%),病毒防护效果持续至少6个月。
2.2同类产品同时具有预防和治疗功能
随着免疫学研究的发展,疫苗的研发和应用逐渐延伸,从传染病扩大到系统性疾病,包括癌症;从预防用扩大到治疗用。
全球范围内,卡介苗主要用作预防儿童结核病的预防接种疫苗,接种对象为出生3个月以内的婴儿或用旧结核菌素试验阴性的儿童。除用作结核病疫苗外,卡介苗还是非肌层浸润性膀胱癌的主要治疗手段。1976年,Morales医生首次将卡介苗直接注入膀胱,治疗复发性浅表膀胱癌获得成功。随后大量的临床观察和研究表明,卡介苗灌注治疗术后残存膀胱癌的完全缓解率为50%~90%(平均70%),有效降低了膀胱癌复发率,推迟癌症复发和病情进展。尽管卡介苗预防结核病和治疗膀胱癌的用法不同,但都是通过激活机体的免疫系统,增强主动免疫功能发挥药理作用。
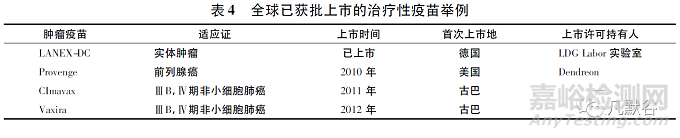
除卡介苗之外,全球已有多款治疗性疫苗上市,见表4。2010年4月29日,美国FDA批准Provenge疫苗,是以患者自身免疫细胞制成的前列腺癌免疫疗法,开创了癌症免疫治疗的新时代。疫苗可以是治疗用或者预防用,但是本质上是同一类通过主动免疫产生免疫应答的产品。而目前的生物制品分类方式,将产品的预防用和治疗用功能截然分开。
2.3基于免疫学原理看生物制品的创新问题
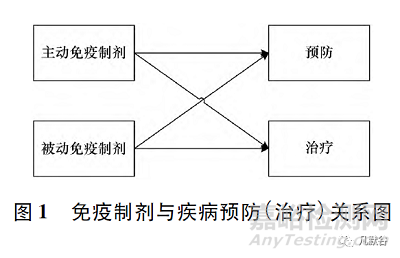
人工免疫是指人为地使机体获得适应性免疫,包括2种:人工主动免疫是用疫苗接种机体,使之主动产生适应性免疫应答,从而预防和治疗疾病的措施;人工被动免疫是给人体注射含特异性抗体等制剂,使之被动获得适应性免疫应答,以治疗或紧急预防疾病的措施,见图1。目前生物制品的注册分类方式,主动免疫制剂纳入预防用生物制品,而被动免疫制剂纳入治疗用生物制品。
2.3.1被动免疫制剂和主动免疫制剂都可以用于预防疾病
一个世纪以来,被动免疫制剂已成功应用于狂犬病、破伤风和乙型肝炎等一系列疾病疑似暴露的暴露后预防或感染后的辅助治疗。随着科学技术飞速发展,以健康人预防用长效单抗为代表的被动免疫制剂可能为某些传染病的预防提供干预手段,如RSV、疟疾等。因为这些疾病疫苗研发难度大,目前仍缺乏有效预防手段,造成严重的疾病负担。世界卫生组织为此推出了一系列单抗预防疾病的理想产品特性清单。
尽管被动免疫保护效力较短,但用单抗进行被动免疫相较于疫苗进行主动免疫有以下优势:①被动免疫起效快,即时就会有保护作用。而且在免疫抑制的个体中仍具有保护效力,这些个体往往疾病感染风险最高。②新技术可以简化单抗的生产或给药,如延长抗体的半衰期或注射编码单抗的mRNA。③生产过程通用,原则上可以在短时间快速开始生产,在疫情暴发时迅速提供产品。
2.3.2被动免疫制剂和主动免疫制剂都可以用于治疗疾病
免疫治疗是指利用免疫学原理,针对疾病的发生机制,人为地干预或调整机体的免疫功能,达到治疗疾病目的所采取的措施,包括主动或被动免疫治疗。
美国批准抗前列腺癌治疗性疫苗上市,促进了抗肿瘤治疗性疫苗的研发,治疗性疫苗已然成为医药生物技术与产业化的热点之一。治疗性疫苗是指机体在感染或发生疾病后,用诱导机体产生特异性免疫或非特异性免疫的方法,防止疾病的发生、发展,或促进已产生疾病的机体恢复健康,多用于慢性感染或肿瘤的治疗。各种治疗性疫苗的作用机制与途径不尽相同,但最基本的理论基础总是围绕如何调节机体的免疫应答,或增强或降低,以达到对所患疾病有不同程度的缓解、控制甚至治愈的效果。
基于免疫学原理,预防疾病的生物制品除了主动免疫制剂,如疫苗类生物制品,还包括以预防为目的的被动免疫制剂,如健康人预防用单抗等新型生物制品。
3、典型国家和地区生物制品分类和注册申请路径
3.1生物制品分类
3.1.1美国的生物制品分类
FDA对“生物制品”定义为:由FDA监管,用于诊断、预防、治疗和治愈疾病和病症,通常是一类大而复杂的分子,并且种类繁多的产品。此类产品通常运用生物技术在生物体中产生,如微生物、植物细胞或动物细胞,并且比小分子药物更难表征。
2003年6月30日,FDA将生物制品评价和研究中心(CBER)审评和监管的一些生物制品转移到药品评价和研究中心(CDER)。CDER对转移的生物制品负有监管责任,两大中心共同促进美国生物制品监管的发展。其中细胞产品,基因治疗产品,疫苗和疫苗相关产品,变态反应原制品,抗毒素、抗蛇毒血清和蛇毒,血液、血液成分、血浆衍生品,人体细胞、组织、细胞-组织产品归属CBER监管。单抗、治疗用蛋白(疫苗和血液制品除外)、免疫调节剂、生长因子、细胞因子归属于CDER监管。
3.1.2欧盟的生物制品分类
在欧盟,生物制品包括3类:使用生物技术生产的生物制品、先进治疗产品(ATMP)和其他生物制品。使用生物技术生产的生物制品申请由EMA的人用药品委员会(CHMP)负责审评,先进治疗产品由先进疗法委员会(CAT)负责评估产品的质量、有效性和安全性,以上2类产品通过集中审批程序获得授权。其他生物制品(如天然衍生生物制品)可以在各个成员国获得国家授权,而不通过集中审批程序。
3.1.3日本的生物制品分类
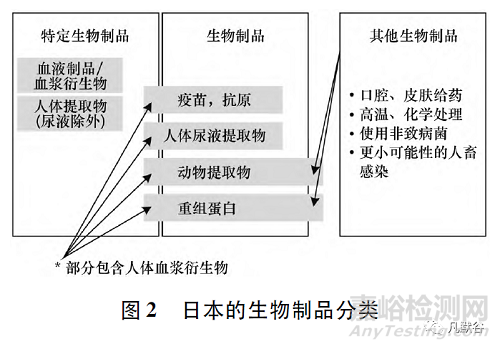
在日本,对生物制品的潜在感染传播风险进行合理科学评估的基础上将生物制品分为特定生物制品、生物制品和其他生物制44品(见图2)。此外,日本将细胞和基因产品等独立出来归于再生医疗产品,进行单独管理,并在修订的《药品和医疗器械法》中增订了再生医学产品监管的部分。
3.1.4生物制品的分类差异
除我国将生物制品分为治疗用、预防用和诊断用进行注册分类外,美国、欧盟和日本并未进行这样划分,见表5。国际上对生物制品分类的考虑因素包括生物制品的生产来源、生物技术的先进性、分子大小和表征复杂性等。欧盟和日本在传统生物制品之外,分别增加先进疗法和再生医学产品的分类。

通常,生物制品注册分类由各国药品监管机构制定用于生物制品上市审评审批,由于各国监管机构对生物制品的分类考量因素不同,因此各国分类各异。但是考虑到部分新型生物制品,包括先进疗法药品等的研发创新,具有区别于传统药物的独特属性,包括治愈潜力、一次性治疗、高额的前期费用、复杂的生产过程和严格的运输条件,因此在生物制品注册分类时纳入“先进性”的考量因素,有助于促进创新,提高ATMPs等新型生物制品的可及性。
3.2注册申请路径
3.2.1美国的生物制品注册申请路径
①新生物制品注册申请路径。生物制品通过生物制品许可申请(BLA)路径上市,其中新生物制品一般通过PH-SA351(a)路径申请,在该路径下需要完整的CMC、非临床以及临床研究数据,即包含完整研发资料的生物制品上市申请。②生物类似药注册申请路径。2010年修订的PHSA中351(k)节中授予了FDA批准生物类似药的权利并且设立了生物类似药简化申请路径。生物类似药只要证明与FDA已许可的生物制品具有生物相似性或可互换性即可通过简化申请路径申请。
3.2.2欧盟的生物制品注册申请路径
欧盟对所有生物制品的注册申请路径分为2类:①完整资料申请路径,欧盟2001/83/EC指令第8条第3款规定,完整资料申请应当提交全面、完整且没有任何研究/试验报告的简化,包括药学(物理化学、生物、微生物)试验、非临床与临床试验结果等在内的申报资料。②生物类似药的简化申请路径,生物类似药申请,即满足2001/83/EC第十条第四款规定的简化申请。简化申请与完整申请相比,在非临床研究以及临床试验部分,可以提交简化的申报资料,或者只需要提交生物利用度研究证明生物等效性即可。
3.2.3日本的生物制品注册申请路径
日本的药品审评注册分类不划分为“化学药品”和“生物制品”,而是以“新药”、“改良型”和“仿制药”(生物类似药)的基本逻辑来划分。具体包括:①含有新有效成分的药品。②新医疗用复方制剂。③新给药途径药品。④新适应证药品。⑤新剂型药品。⑥新用量药品。⑦生物类似物。⑧增加新剂型的药品。⑨类似处方医疗用复方制剂。⑩其他处方药。
依据2005年3月31日,日本厚生劳动省医药食品局发布《关于药品生产销售的批准申请通知》(第0331015号令)。按照药品的创新程度以及申报资料的完整程度,注册分类1类到10类的处方药品,随着其创新难度的降低,其注册申请所要求的资料也逐渐简化。
3.2.4生物制品的注册申请路径差异
从各国生物制品的注册申请路径来看,只有我国把生物制品分为治疗用和预防用2类。美国和欧盟的生物制品注册分类按照“创新”和“仿制”(生物类似药)的原则,将注册申请路径分为完整申请路径和简化申请路径,日本按照药品的创新程度以及申报资料的完整程度将注册分类分成1~10类,见表6。


4、关于生物制品分类体系的思考与建议
4.1建立持续改进的生物制品分类体系
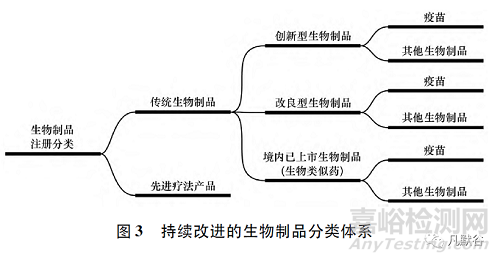
科学的本质在于分类,对生物制品的注册分类进行适时调整是适应生物制品创新和产业发展,紧跟科学技术进步的必然要求。随着科学和技术的发展,生物制品治疗与预防的功能逐渐交叠,未来生物制品的注册分类不适合按照产品类别进行预防用和治疗用的简单功能区分,应当按照国际通行的创新、改良和生物类似药的分类逻辑进行申报资料规定。考虑到我国一直以来对疫苗有单独注册要求,建议新的生物制品注册分类见图3。
4.2区分传统生物制品与先进疗法产品
先进疗法产品包括基因治疗产品、细胞治疗产品和组织工程产品,这些高度复杂的治疗产品无论是生产和给药途径,还是临床疗效均不同于传统药物。一些基因治疗产品可以解决疾病的病因,为患者提供一次性治愈的机会,也有一些细胞治疗产品和组织工程产品是专门为特定患者而生产的,形成个体化定制药物。将先进疗法产品与传统生物制品进行注册区分(图3),有利于监管部门制定一系列监管策略,支持新的先进疗法产品的早期开发,以便促进患者可以更早获得这类产品。
5、结语
随着生物制药技术的飞速发展,全球涌现出一系列的创新型生物制品,如预防用单抗和治疗用疫苗等。目前生物制品的注册分类按照产品类别进行预防用和治疗用的简单功能区分,新型生物制品可能将面临注册分类与实际用途不匹配的问题。建立持续改进的生物制品分类体系,区分传统生物制品和先进疗法产品,有利于生物制品的创新和产业发展,便于监管部门制定监管策略,助力患者更早获得创新产品。