您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-10-12 20:56
本文来源于广东省药品不良反应监测中心整理的注册和备案人在日常工作中常见的问题,依据医疗器械监督管理及不良事件监测相关法规和指南性文件,为注册人备案人提供答疑。
收集的问题不一定全面,后续将不断完善或补充。问题的答疑如跟现有法规冲突的,以法规解释为准。如果相关问题法规有变化的,广东省药品不良反应监测中心将对答疑的内容作相应调整。
国家医疗器械不良事件监测信息系统
登录入注册问题
问国家医疗器械不良事件监测信息系统登陆地址是什么?
答http://maers.adrs.org.cn
问国家医疗器械不良事件监测信息系统如何注册?
答1.2019年前已在国家医疗器械不良事件监测信息系统注册过的持有人,可以使用原来的帐号登录,若无法登录,请联系市监测中心。
2.未在国家监测系统注册过的用户,需要在监测系统注册帐号。在系统登录页面中,单击“用户登录”区域中“注册”链接,页面跳转至用户注册页面。在“用户类型选择”单选按钮组中选择“用户类型”为“持有人”,页面中显示持有人用户注册表单。持有人用户注册页面如下图所示:
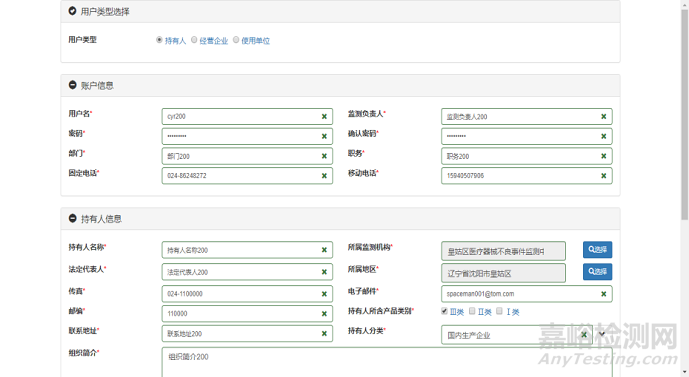
图1 持有人用户注册页面
在持有人用户注册页面中,用户根据提示信息填写注册表单。填写完毕后单击页面右下角区域【提交】按钮,系统自动将注册信息提交至所在地省级监测机构,并提示“您的注册信息已经提交成功,请您耐心等待审核结果,系统会在5天内通过电子邮件方式通知您!”。单击【返回】按钮,页面跳转至系统登录页面,完成用户注册操作。帐户注册经审核通过后,注册人须在系统完善企业信息并录入医疗器械产品信息。
问在监测系统中注册时填错了用户身份类型,应该如何处理?
答如果持有人在注册时选错身份类型,可以联系市监测中心,或重新注册。
问如果企业既是生产企业又是经营企业,在监测系统中该选用何种身份注册?
答企业既是生产企业又是经营企业,应分别以生产和经营企业身份注册帐号,但用户名不能相同。
监测系统信息维护问题
问机构信息变更后要怎么做?
答1.注册人名称变更的,需要填写《系统平台用户信息变更申请单》,提交市级监测中心。
2.修改注册人名称变更以外信息的,在系统“机构修改”处点击“新增”,修改相应信息即可,修改完毕后保存提交审核。
问注册人如何在国家监测系统录入产品信息?
答 1.所有在产、在售、在用的医疗器械注册证或备案证都须录入国家监测系统。以附件的形式上传产品注册证或备案证扫描件、说明书与技术标准,扫描件仅支持Word和PDF格式,文档大小在60M内。一类医疗器械的有效期统一填至3000-01-01。
2.对于已延续注册的产品,现有注册证号与旧注册证号不一致时,若旧注册证号对应产品并未超过有效期且仍在市场上流通使用,则需要把旧注册证号录入在“曾用注册证号”位置。注册证号有变更的需以附件提交注册证扫描件和变更文件(扫描在一起)。
问注册人无法在国家监测系统录入注册证号如何处理?
答1.录入产品信息时曾用注册证号无法手动录入,需要点击文本框右侧“生成”键,在子页面中勾选曾用注册证号的格式校验后点击“生成注册证编号”,系统会给出注册证号参考格式,将曾用注册证号在此处录入,点击“确定”即可。
2.注册证号中带有“(更)”、“(补)”字样的,请省去该字样,只录入前面的信息即可。
问在国家监测系统录入产品信息且经审核后,如果发生变更或发现录入有误应如何维护信息?
答1.注册证号或曾用注册证号录入错误的无法修改,需要停用处理,重新录入。
2.非注册证号错误,需要修改其他错误信息的,在基础数据管理页面点击查询,选中需要修改的产品信息,点击“修改”,对照变更文件修改相应信息(如规格、型号、产品名称等),同时把变更文件和现用有效注册证扫描在一起作为一个文档替换掉之前的注册证文件。扫描件无需删除原来的扫描件,重新上传覆盖即可。
3.产品办理了延续注册的,有效期发生变更但注册证号未变更的,则在监测系统中选择该注册证号,点击换证延续中的“修改”,打开修改医疗器械信息页面,在注册批准日期列表区域,点击第一列“+”号,新增一条延续记录,录入延续起始日期和延续有效期,系统会自动计算延续年限,填写完成后并上传相关附件后点击保存按钮,即完成产品不换证延续。
4.产品办理了延续注册,注册证有效期及证号发生变更的,选择需要换证延续的产品,点击换证延续按钮,打开“医疗器械信息换证延续”,在该页面填写换证产品信息,填写完成并上传附件后点击保存提交省监测中心审核。
问应急医疗器械注册证或备案证有效期满后再正式注册的,如何在监测系统维护信息?
答1.应急医疗器械注册证或备案证有效期满后正式注册或备案的,产品注册证或备案证有效期内在国家监测系统中若未收到不良事件报告,注册人可向市级监测机构申请退回该产品注册证或备案证的审核,并由注册人自行删除该条产品信息,然后由注册人重新录入正式注册或备案的产品信息。
2.应急医疗器械注册证或备案证有效期内在国家监测系统中若已收到不良事件报告,后来又正式注册或备案的,注册人需向国家药品不良反应监测中心提出书面申请,由监测系统工程师修改应急医疗器械产品信息的有效期(修改为正式注册证或备案证的有效期,二、三类医疗器械还需修改为首次注册)。
问产品注册证到期后未延续,国家监测系统中的产品注册证信息能否删除?
答 产品注册证或备案证已注销或已失效未延续的,注册证信息不能删除。
问国家监测系统的“迁入”、“迁出”是什么意思?
答当产品的持有人或注册代理人有变更时,该产品须迁出到新的持有人或注册代理人的帐户,新的持有人或注册代理人需在监测系统中办理迁入手续,产品的迁出和迁入均在监测系统完成。产品迁出时,若涉及多个曾用注册证号,需一并迁出。
医疗器械不良事件报告与处置问题
问医疗器械不良事件的报告原则是什么?
答医疗器械不良事件应当遵循可疑即报的原则。
问医疗器械不良事件报告的基本要求是什么?
答医疗器械不良事件报告的基本要求是真实、及时、准确、完整。
问如何在国家监测系统上报医疗器械不良事件?
答1、境内个例报告上报
注册人用户登录系统后,单击系统菜单中【个例不良事件管理】-【个例上报(持有人)】菜单项,进入个例上报(持有人)页面。单击【新增】按钮,弹出新增页面;选择需要修改的报告信息,单击【修改】按钮,弹出修改页面;选择需要删除的报告信息,单击【删除】按钮,完成报告删除操作;选择需要提交的报告信息,单击【提交】按钮,系统自动将报告信息提交至所在地市级监测机构,完成报告上报操作;选择需要导出Excel文件的报告信息,单击【导出】按钮,完成报告导出操作;选择需要查看的报告信息,单击【查看】按钮,弹出查看页面。个例上报(持有人)页面如下图所示:
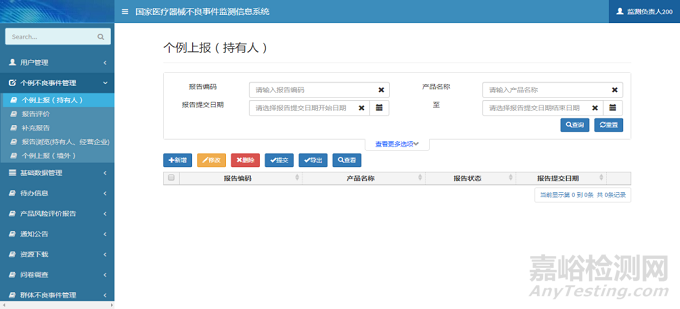
图2 个例上报页面
在新增页面中,用户根据提示信息填写报告信息表单。单击【保存】按钮,完成报告信息保存操作;单击【保存并提交】按钮,完成报告信息保存操作,同时系统自动将报告信息提交至所在地市级监测机构。个例上报(持有人)新增页面如下图所示:
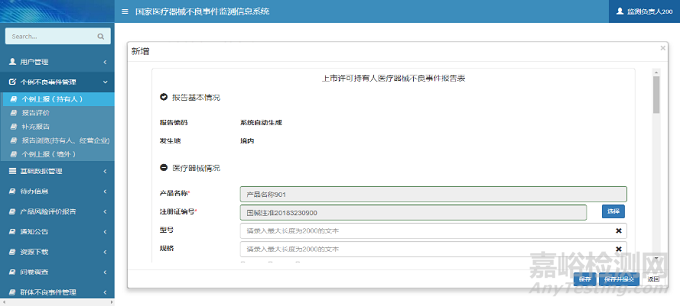
图3 个例上报新增页面
在修改页面中,用户可以维护报告信息。单击【保存】按钮,完成报告信息保存操作;单击【保存并提交】按钮,完成报告信息保存操作,同时系统自动将报告信息提交至所在地市级监测机构。个例上报(持有人)修改页面如下图所示:
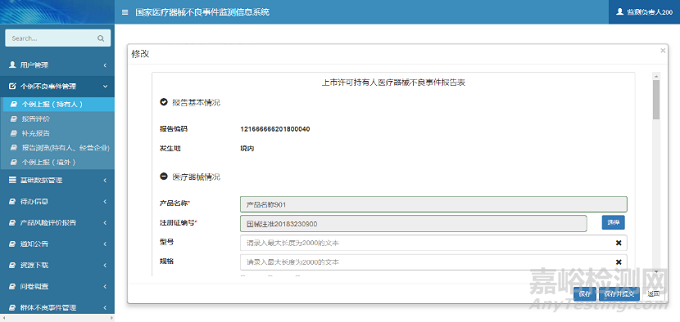
图4 个例上报修改页面
2.境外个例报告上报
注册人用户登录系统后,单击系统菜单中【个例不良事件管理】-【个例上报(境外)】菜单项,进入个例上报(境外)页面。单击【新增】按钮,弹出新增页面;选择需要修改的报告信息,单击【修改】按钮,弹出修改页面;选择需要删除的报告信息,单击【删除】按钮,完成报告删除操作;选择需要导出Excel文件的报告信息,单击【导出】按钮,完成报告导出操作;选择需要查看的报告信息,单击【查看】按钮,弹出查看页面。个例上报(境外)页面如下图所示:
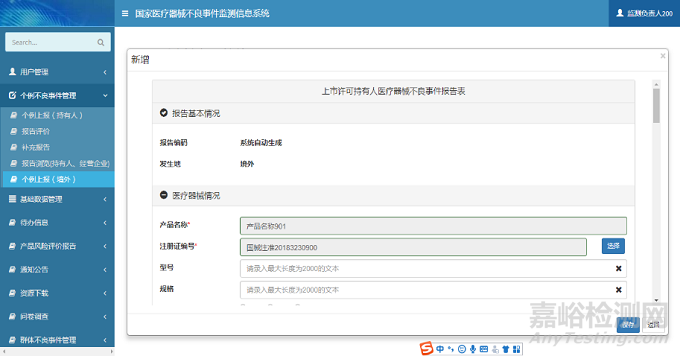
图5 个例上报(境外)页面
在新增页面中,用户根据提示信息填写报告信息表单。单击【保存】按钮,完成报告信息保存操作。个例上报(境外)新增页面如下图所示:
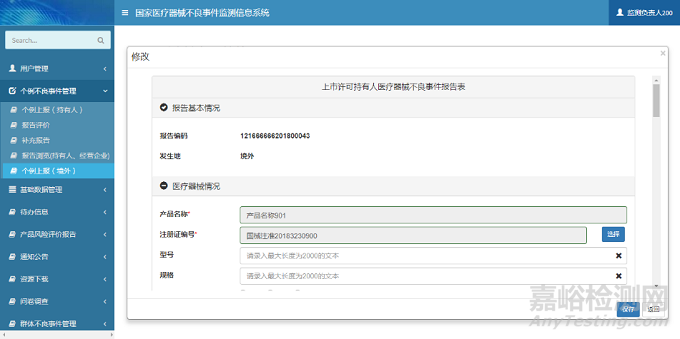
图6 个例上报(境外)新增页面
在修改页面中,用户可以维护报告信息。单击【保存】按钮,完成报告信息保存操作。个例上报(境外)修改页面如下图所示:
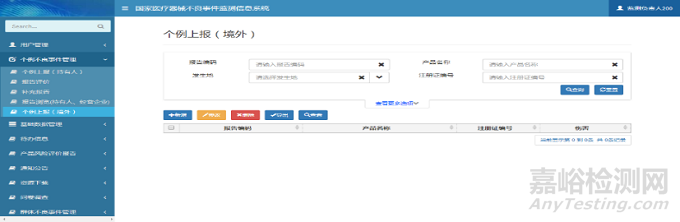
图7 个例上报(境外)修改页面
问注册人如何评价个例报告?
答1.注册人报告或者通过系统获知医疗器械不良事件后,应当按要求开展分析和评价工作,导致死亡的事件应当在30日内,导致严重伤害、可能导致严重伤害或者死亡的事件应当在45日内向注册人所在地省级监测机构报告评价结果。对于事件情况和评价结果有新的发现或者认知的,应当补充报告。
2.通过对个例事件的评价,认为产品可能存在风险的,应当对产品进行风险评价,在不良事件调查核实的基础上,深入分析监测数据、产品检验情况和其他风险信息等资料,必要时召开专家讨论会,在计划时限内通过系统提交评价报告,并根据评价结果采取必要的风险控制措施。
问持有人在国家监测系统评价个例报告时,经分析不需要开展产品风险评价的个例报告,误操作勾选为需要开展产品风险评价,怎么办?
答个例不良事件报告评价时,只要勾选为需要开展产品风险评价,就应按系统提示的要求录入相关信息并及时提交。
问诊断试剂,特别是早筛诊断试剂,不大可能造成人体伤害或死亡,那么诊断试剂检测结果不准(如假阳性)是否属不良事件范畴?
答根据不良事件的定义,诊断试剂出现假阳性或假阴性,可能出现影响诊断、延误治疗等可能导致人体伤害的后果,属于不良事件。
定期风险评价报告问题
问报告期内未收到不良事件报告,或数据汇总期内未生产、未销售,是否仍要撰写定期风险评价报告?
答报告期内未收到不良事件报告,或数据汇总期内未生产未销售,仍需在规定的时限内撰写定期风险评价报告。
问撰写定期风险评价报告的基本要求是什么?
答定期风险评价报告应按时提交;数据应真实、完整,结合监测数据,深入分析风险,全面反映报告期内的风险情况;发现风险趋势,评价结论准确客观;不得填写虚假数据,隐瞒风险;存档备查(一类及延续产品)。具体要求参见《定期风险评价报告撰写规范》。
问定期风险评价报告的数据汇总时间有什么要求?
答 医疗器械首次获得批准注册,数据起始日期应与取得注册证明文件的时间一致,以起始日期后每满一年的日期为数据截止日期。
问定期风险评价报告的附件需要上传哪些文件?
答《定期风险评价报告》的附件包括:定期风险评价报告正文全文,医疗器械注册(或备案)批准证明文件、产品使用说明书(操作手册)、参考文献、其他需要提交的资料。
问应急医疗器械是否需要撰写定期风险评价报告?
答应急医疗器械无需撰写定期风险评价报告。应急产品注册证或备案证有效期满再正式注册的,须按首次注册的要求撰写定期风险评价报告。
问定期风险评价报告是否可以补报?
答监测系统允许持有人补报定期风险评价报告。因为1号令是2019年1月1日开始实施的,故最早的补报报告数据汇总期为2018至2019的那份(即数据汇总期跨过2019年1月1日那份报告),2018年以前的无需补报。大家可通过学习网文《一文读懂〈定期风险评价报告撰写要点〉》了解更多具体要求。

来源:广东省药品不良反应监测


