您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-09-08 11:31
什么是UDI系统,我们为什么需要它?
UDI(unique device identifier),即医疗器械唯一标识。
美国国会指示FDA希望建立一个UDI系统,通过其分配和使用来充分识别医疗器械。
建立UDI系统的目的有以下几点:
1.便于快速、准确地识别设备
2.允许访问有关设备的重要信息
3.在电子健康记录、临床信息系统、索赔数据来源和注册中提供标准和清晰的方法来记录设备使用情况
为达到这些目的,需要满足以下四步:
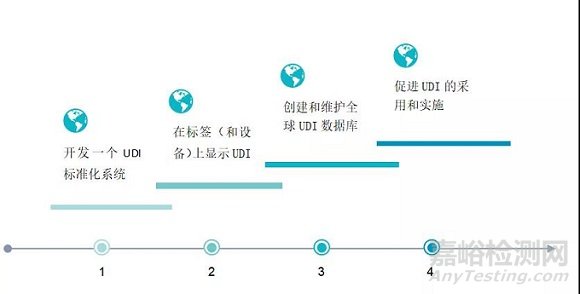
FDA已于2013年9月24日公布了UDI Final Rule。
现在,整个医疗器械行业需要遵守UDI规则。器械标签和器械包装上都要有UDI,在某些情况下,器械本身也需要UDI,除非有例外情况或FDA批准的替代方案。关于每个设备的数据需要提交给全球UDI数据库,简称为GUDID。
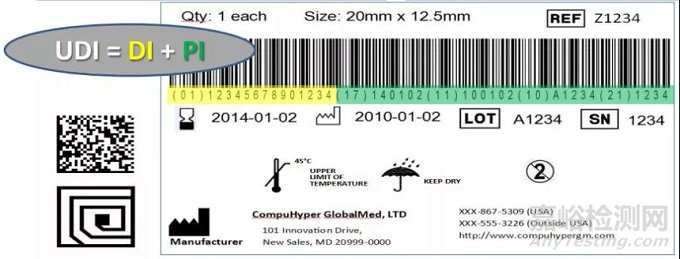
02UDI组成结构
UDI,包括DI(device identification)和PI(production identification)两部分。DI为固定的产品信息(如厂商和医疗器械的型号等),PI由制造商自己定义,一般包含产品批号、序列号、有效期和生产日期等。
03Labeler要对UDI负责
Labeler负责符合UDI要求。Labeler通常是制造商,也可能是合同制造商、自有品牌经销商等。Labeler需要与至少一个被认可的发证机构合作并使用其规则来构建它们的UDI。目前FDA认可的签发机构共有三家,GS1,Health Industry Business Communications Council (HIBCC)和ICCBBA。
04UDI要求的遵守日期
UDI要求的遵守日期主要基于器械分类,在7年的时间内分阶段进行,高风险器械的遵守日期较早。以下是关键的合规日期。
|
器械 |
UDI要求的遵守日期 |
|
公共卫生署(PHS)法案许可的3类(包括3类 I/LS/LS1)器械 |
2014年9月24日 |
|
可植入的,生命支持和生命维持的器械(2类,1类和未分类) |
2015年9月24日 |
|
2类(I/LS/LS1除外) |
2016年9月24日 |
|
1类或未分类(I/LS/LS1除外) |
2018年9月24日 |
|
直接(在器械上)标记(DM) |
2020年9月24日 |
I/LS/LS1:可植入的,生命支持和生命维持的器械
所有医疗器械必须在适用的遵守日期前符合UDI规则的要求,除非FDA批准了例外或替代方案,如:
· I类cGMP豁免器械
· 单独的一次性器械,以单一包装出售和储存,直到取出使用
· 仅用于非临床用途的器械或IDE
· 仅供美国出口的器械
· 便利性工具包中的单个器械
· 在遵守日期之前包装和标签待售的器械在遵守日期之后三年内可以不符合UDI要求
总结UDI的基本规则:UDI在每个设备标签和设备包装上都是必需的,在某些情况下在设备本身上,除非有例外情况或被FDA批准的替代方案。设备的关键信息必须提交给GUDID。所有医疗器械必须在适用的遵守日期前符合UDI规则的要求,除非FDA批准了例外或替代方案。

来源:奥咨达医疗技术服务


