您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-09-16 08:51
2020年9月10日,国家药品监督管理局药品审评中心再放大招,官网发布《临床试验期间生物制品药学研究和变更技术指南》征求意见稿,征求意见截止时间2020年10月9日。一石激起千层浪,此变更技术指南对生物制品行业具有深远的意义。此次的《临床试验期间生物制品药学研究和变更技术指南》征求意见稿的发布,突破性地提出了临床试验期间变更的实际申报要求,弥补了生物制品临床试验期间变更监管的空白,有助于确保受试者安全,推动协调生物制品临床试验及上市注册进程,同时也为生物制品研发者提供临床期间药学的研发思路、方法及阶段性要求,临床试验期间生物制品药学变更如何合规是当前生物制品上市许可持有人(MAH)最迫切关心的问题之一。
一、对标国际标准,高标准的中国造生物制品
为规范临床试验期间生物制品药学研究及其变更,满足阶段性临床试验用样品的基本要求,保护临床受试者的健康,CDE以现有国家颁布的相关法律、法规及技术指导原则为基础,立足国内生物制品注册申报现状,借鉴发达国家的先进管理理念,结合国内外相关法规和技术要求,本着科学性、可操作性、前瞻性和先进性相结合原则,经过前期调研、初稿撰写、专家座谈会讨论以及部门技委会讨论后,形成了《临床试验期间生物制品药学研究和变更技术指导原则》,本指南引用主要国内外参考文献和指导原则,清单如下:

二、临床期间变更覆盖范围和前提条件
药物临床试验目的是确证试验药物的疗效与安全性而开展的系列性研究。临床试验期间变更管理属于药品全生命周期管理的一部分,临床试验期间变更研究工作越系统,生产过程中积累的数据越充分,对上市后的变更研究越有帮助,CDE首先从顶层设计层面提出了临床研究期间药学变更的覆盖范围和前提条件:
(1)【覆盖范围】
"临床试验期间"覆盖申办者从临床试验申办者获得临床试验默示许可后到提交上市申请前的整个阶段。
(2)【前提条件】
临床试验期间生物制品药学研究具有阶段性、渐进性特征,研究的深度和广度在很大程度上取决于临床试验的阶段,并与后续拟开展临床试验安全、有效和质量可控性的相关要求相衔接。前提条件是以不增加临床受试者安全性风险为前提,使前期研发数据能够支持后期临床试验的开展,并为生物制品最终上市提供充分的支持性数据为目标。
三、不同品种临床研究期间药学变更评估的原则
由于不同类型生物制品差别极大,临床期间变更情形复杂多样,仅反映了当前我们对生物制品临床试验期间药学研究和变更问题的观点和认知,内容将随着科学的发展和行业有关人士对生物制品临床试验期间研究和变更认识的深入及时修订与更新,不同类型的生物制品临床期间药学研究和变更的侧重点不同。
(1)创新型生物制品:
创新型生物制品是指境内外均未上市的生物制品,在早期临床阶段,通常建议应建立与产品安全相关的过程控制(包括工艺参数和验收标准)及关键步骤的可接受标准。非安全性相关的过程控制、可接受标准和行动限,应随着研发不断地完善;在确证性临床时,鼓励采用商业化生产规模的工艺和标准。
(2)已上市生物制品
已上市生物制品相关研究成熟度和基础条件较高,成药确定性通常较强,临床试验的风险性能够通过扩充临床前研究使其控制在相对较低的水平,临床期间药学研究和变更应在具备商业化生产的规模和基础上,重在局部调整优化和衔接性研究,提前为上市做验证准备。特别是在减免阶段性临床试验及人体数据要求时,不仅应提供确保受试者安全性的药学研究,还应补充支持产品有效性评价的药学研究。
(3)改良型生物制品
改良型生物制品是指对境内或境外已上市生物制品进行改良,使新产品的安全性、有效性、质量可控性有改进,且具有明显优势的生物制品,改良型生物制品介于上述两类生物制品之间,临床申办者可根据品种的具体改良特点及改良的成熟度与安全性等信息,结合临床试验期间研发策略及创新型、已上市生物制品阶段性要求,适时完成各阶段的药学研究和变更工作。
(4)特殊品种
特殊品种,如基因治疗、细胞治疗等先进疗法生物制品,评估变更产生的影响可能相对困难,临床试验期间发生变更进行可比性研究时,建议关注共用的产品工艺、原材料和质量特性等方面,结合个体差异,综合评估变更引入的影响和风险。
四、临床试验期间药学变更后CTD格式资料如何衔接?
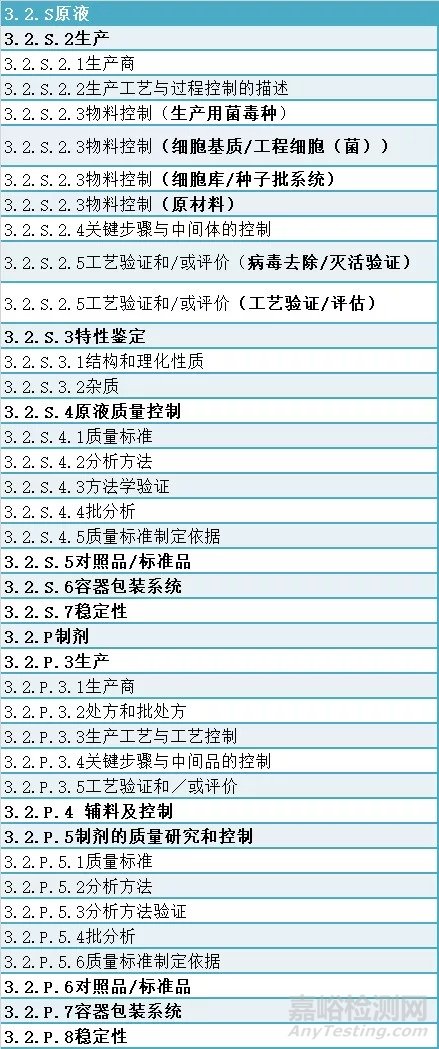
推广CTD格式是加速中国造生物制品申报与世界接轨的桥梁,为更好地进入国际市场做好铺垫,同时,也是为了鼓励国内企业开展新药研发,进一步规范注册资料申报格式和内容,为配合2020年7月1日起实施《药品注册管理办法》及《生物制品注册分类及申报资料要求》,CDE要求物制品申报资料自2020年10月1日起实施CTD格式申报。临床试验期间原液和制剂中相关研究内容发生变更时,可以参照下表内容顺序起草补充申请资料,衔接CTD格式申报资料。
参考文献
[1]www.cde.org.cn

来源:CPhI制药在线


