您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-07-06 10:24
近年来,随着药物研究的不断深入以及杂质研究要求不断提高,杂质的分析技术以及研究方法正发生着重要的改变。在对杂质建立分析方法时,清晰的杂质研究过程是方法建立的基础,而且选择合适的分析技术也至关重要。
本文提出了杂质来源分析的重要作用,同时重点探讨了杂质研究过程中分析技术的发展,尤其在结构鉴定中质谱技术的发展,与此同时,根据国外毒性杂质研究的指导原则,明确了杂质毒性研究的方法。
1、杂质的来源分析
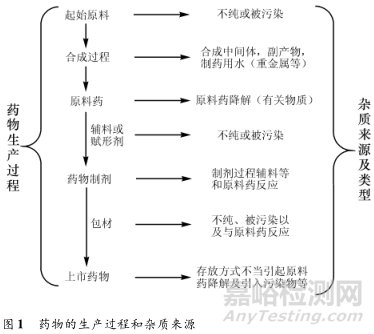
药物中的杂质可能来源于药物生产以及销售等各个环节(图 1)。根据 ICH指导原则可将药物杂质分为有机杂质、无机杂质、残留溶剂以及其他杂质。本文主要针对有机杂质进行探讨。
对药物杂质研究时引入“质量源于设计( Quality byDesign,QbD)”的理念,可在药物生产之前根据具体工艺的合成机制、起始物料及各中间体的基本结构,初步勾画出产品的杂质谱。
杂质来源分析是制定药物杂质控制策略的基础,尤其是在对毒性杂质来源分析时,应分析所有合成和生产工艺中的试剂、中间体、副产物,推测可能产生的潜在杂质以及分析实际存在的杂质。在原料药合成结束后,药物的活性化合物虽然经过毒性分析已不含有“警示结构”(alerting structure),但是在生产过程中使用到含有警示结构的化合物则还需考虑其遗传毒性。
2、杂质的研究方法
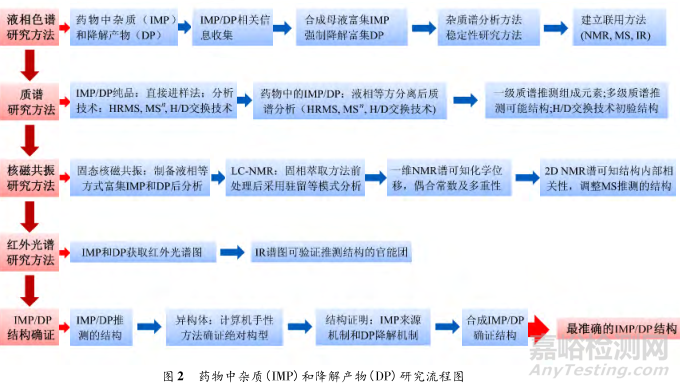
在药物研发过程中,药物杂质的分析是关键。因此,在杂质研究中清晰的杂质结构研思路(如图 2)以及合适的杂质分析技术可极大地缩短杂质研究时间,推动着药物研究的快速发展。
2.1 杂质前处理技术
杂质的前处理是伴随着药物活性成分前处理而存在的,然而药物中杂质的含量低且其结构与主成分差异较大,因此常规药物活性成分的前处理和检测方法(如初始流动相溶解后直接进行 HPLC-UV 分析)并不一定适用于药物杂质,应针对不同的样品选择不同的前处理技术。
2.1.1 检测灵敏度低的样品
对检测灵敏度低的样品通常使用衍生化的前处理方式,比如引入生色团产生紫外响应,或增加易离子化基团增加离子化效率等。
虽然常规衍生化方式能够满足日常检测的需求,但是为了实现对低浓度的基因杂质进行快速筛选和定量,可对传统的衍生化试剂进行改变以增加其专属性和灵敏度,也可使用气-固衍生化来弥补液-固衍生化的不足。
2.1.2 低浓度的杂质
低浓度杂质前处理方法的选择根据其杂质类型所决定,如降解产物利用强制降解等方法提高降解物的浓度等,但是常规的降解方法往往会引入其他杂质,因而会干扰特殊杂质的杂质谱研究,为了得到单一的杂质研究机制,Ueya-ma 等提出了一种新型的固体药物氧化降解平台,该平台排除了常见氧化方式(例如 H2O2 主导)引起的水解、溶剂解或热效应等,可用于氧化降解机制的特异性研究。
2.1.3 易污染仪器的样品
不同仪器有不同的使用条件,因此对复杂样品进行前处理工作能够延长仪器的使用寿命,例如质谱检测器不能使用含有非挥发性盐的流动相,因此在建立液质联用条件时可利用二维液相色谱技术在第一维将各峰进行分离并将样品保留至样品环中,第二维液相使用质谱可接受的流动相以及脱盐柱来洗脱样品环中的样品从而实现了被分析物“脱盐”来保护质谱。
2.2 杂质分离技术
杂质在药物中含量较低,利用直接测定法无法实现杂质的定性定量分析。因此,应对杂质进行分离以获取杂质的单一成分,从而实现对杂质的检测。近年来,液相色谱技术以及超临界流体技术发展较为迅速。
2.2.1 液相色谱技术
高效液相色谱技术(HPLC) HPLC 作为最传统的方法在杂质分离中仍然使用得最多。通过各种色谱柱技术以及联用技术能够对绝大多数化合物实现分离、分析。为了解决与紫外检测器联用灵敏度低的问题,新型二维 高 效 液 相 色 谱 (2D-HPLC)利用了液相色谱技术分离和富集的特点提高了对低浓度杂质的检测能力。
目前,HPLC 无论采用正相洗脱还是反相洗脱都需消耗大量的有机溶剂,这对环境带来较大的污染,因此提出以水或其他环境友好的试剂为主要洗脱溶剂的新型液相色谱技术正得到广泛的研究。超高效液相色谱技术(UHPLC) 为了应对药物研发的需要,UHPLC 作为一种快速分离的色谱技术已经出现在各种药物的开发上。
Dong 等根据UHPLC 的使用方法,讨论了如何针对不同样品建立稳定的 UHPLC 方法。手性分离方面,UHPLC目前并没有大量小于2 μm 粒径的商品化手性色谱柱,但是利用传统的手性流动相添加法也可实现对手性杂质进行分离。
在实验室建设方面,分析实验室目前仍以 HPLC 为主要研究仪器,但为了获得类似于UHPLC 分离效能,许多科研工作者通过优化 HPLC系统同时使用核壳型色谱柱实现了快速分离。
2.2.2 超临界流体色谱(SFC)
以液态CO2 为主要流动相的SFC 技术,由于其与紫外联用检测灵敏度低,其发展一直非常缓慢,但质谱检测器(MS)的普及以及环境友好型社会的需要,使 SFC 能用于分离手性杂质。虽然 SFC 与质谱联用技术用于杂质分析已有相关报道 ,但商品化的 SFC-MS联用仪器并未大量出现。
2.3 杂质制备与纯化技术
为实现对杂质的定量及定性分析,需要获取高纯度单一成分的杂质,然而杂质在药物中含量较低,利用分析型液相色谱技术制备杂质需要消耗大量时间,利用制备型液相色谱技术等方法可提高高纯度杂质的获取速度,加快杂质研究工作。
2.3.1 制备型液相色谱技术和制备型SFC技术
经纯化的杂质可获得更高质量的图谱,但药物中杂质含量低的问题一直制约着杂质单体的获取速度。利用液质联用方法可获得杂质的来源与简单结构,再利用强制降解 、结晶母液或直接合成等方式可制备出杂质单体制备技术上。
为了克服常规一维制备型液相色谱技术以及制备型SFC 色谱技术的方法建立困难、制备时间较长等缺点而提出制备型的二维液相色谱技术(Prep 2D-LC)以及二维 SFC 色谱技术已得到越来越多的应用。
Zhang 等提出了一种新型的 Prep 2D-LC,该仪器首先通过一维液相色谱对样品进行初步分离,并用质谱相对分子质量监控和中心切割的方式将目标物保存在样品环(Sample loop)中,再利用在柱洗脱(at column dilution)的方法将样品环中的样品进样至第二维液相色谱中,第二维液相色谱通过使用与第一维相同或者不同的流动相对样品进行进一步分离同时使用质谱监控方法即可得到更高纯度的目标物。
这种新方法不仅替代了一维制备型液相色谱,同时解决了传统制备 2D-LC 系统高压以及峰展宽的问题。类似于 2D-LC,该课题组也提出了 2D-SFC 制备技术以用于手性和非手性化合物的筛选 。
2.3.2 其他制备技术
药物中杂质含量低,利用液固吸附分离的方式会造成杂质损失,延长制备时间。逆流色谱技术 ( counter-current chromatogra-phy,CCC)通过液液萃取方式使杂质吸附达到最低,样品重现性能够达到 100%,而且有报道将制备液相色谱技术和 CCC 进行对比,结果表明 CCC对溶解度低的样品的上样能力和高通量能力都优于制备液相色谱。
与 CCC 类似的离心分配色(centrifugal partition chromatography,CPC),利用流体静力分配方式实现对药物的纯化作用,但CPC 在使用中会出现固定相流失以及流速不稳定脉冲的问题,Amarouche 等 通过引入顺流洗脱(co-current elution)方法解决了上述问题并成功地对不溶性环孢素 A 进行了纯化。
为了实现快速制备与纯化,通过空气加压加速液固分离的快速质谱(flash chromatography,FC)也不失为一种大量制备化合物的方法。
2.4 杂质检测技术
虽然 HPLC-UV 技术可对大部分药物杂质进行定量分析,但由于紫外检测灵敏度低而无法实现对极微量杂质的准确定量。然而,质谱技术拥有高灵敏度和高分辨率等优点,近年来由于其具有卓越的定量和定性分析能力已得到了快速的发展。同时,减少样品消耗量也是不断推动核磁共振技术的发展动力之一。
2.4.1 质谱技术(MS)
定量分析
质谱技术可作为紫外无响应杂质的一种替代定量手段,同时因其具有较高的检测灵敏度,能够对紫外单波长检测无法定量的痕量杂质得以准确测定。但在对某些特殊杂质测定时,由于离子化能力弱仍需要通过衍生化或在流动相加入碱金属离子等方法以获取质谱响应 。
常规 MS检测器由于分辨率的制约导致对痕量杂质的定量准确性不高,而高分辨率质谱(HRMS)能将被分析物的荷质比(m/z)检测相对误差降到1 ×10-6~2 ×10-6,在选择离子扫描定量中提高了对痕量分析物的定量准确度。
HRMS高分辨率的另外一个优点是质量区分更为准确,这可用于区分相对分子质量相近的多种化合物,利用此优势和 UHPLC联用 能 在 短 时 间 内 实 现 对 多 种 杂 质 的 分 离定量 。
结构鉴定
对杂质单体进行结构鉴定耗时长,因此在药物的杂质谱分析中使用液质联用的方法可对杂质结构进行快速鉴定。该方法是以一级质谱确定的分子离子峰进行二级质谱碎裂分析。
然而,质谱分辨率的不足常使母离子的 m/z 判断不准,从而造成杂质的元素组成不明确,同时二级碎片信息量不足也阻碍了对杂质结构的进一步解析。因此,HRMS、多级质谱(MS n )以及氢/氘(H/D)交换等技术以其各自的优点能够对杂质结构做出准确的解析。
质谱分辨率的提高增加了相对分子质量信息的准确性并可准确预测元素组成,同时利用质谱内置软件或其他计算软件 可计算出不同分子式得分高低。质谱分辨率提高也有区分不同同位素的功能,例如相对分子质量为 500 左右的分子,只有分辨率达到 400000 才能将质谱图中的 34 S 和 37 Cl两种同位素峰分离开。
傅里叶变换离子回旋共振质谱(FT-ICR-MS)一直引领着质谱的高分辨率,因而在新杂质准确分子质量的检测上有独特的优势,Awasthi 等
利用 FT-ICP-MS 准确测定了依立诺克丁中主药及其降解产物的准确分子质量,相对分子质量的相对误差小于1 ×10-6。
然而,FT-ICP-MS 的维护成本高以及扫描速度慢等缺点限制了其在杂质定性中的应用。配备有傅里叶转换仪器同时将磁场替换为电场的 Orbitrap 质谱结合了 FT-ICP-MS 高分辨率以及飞行时间质谱仪(TOF-MS)高灵敏度的特点已经越来越多应用于杂质的研究。
在分子结构分析上,多级质谱(MSn ),如离子阱质谱(IT-MS)等,较 MS/MS 获得更加丰富的碎片,同时能够获取中性丢失分子,使得结构解析更为方便。多级质谱一般和高分辨质谱串联,不仅能得到准确的碎片离子峰,同时获得清晰的裂解途径,使得对复杂分子解析更为方便。
在对磺达肝素钠中的戊多糖杂质进行结构鉴定时,Smith 等使用线性离子阱和 Orbitrap 质谱串联在负离子模式下对硫酸戊多糖进行 MS10 分析,得到了硫酸戊多糖的完整裂解途径,确定了其结构。
作为一种对质谱改进的新技术,H/D 交换技术是利用 H 及其同位素 D 在原子质量上的差异实现对分子中活泼氢个数的测定。该质量差异不仅能用于母离子官能团的判定,同时在多级质谱中对比交换前后碎片离子峰质量差异可了解分子中含活泼氢官能团的裂解路径,以对杂质结构做出佐证。
利用强制降解实验对药物中的降解产物进行研究时,强氧化等方法得到的降解产物常出现新的官能团,利用在线的 H/D 交换和多级质谱分析并通过与药物活性成分质谱图的对比能实现对含有活泼氢的新官能团类型以及位置的确定;对相同分子质量的烯醇互变杂质分析时利用 H/D 交换的方法能非常方便地判别出烯醇杂质的结构。
H/D 交换的方法将流动相中 H2O 更换成 D2O,使被分析物在流动相洗脱过程中实现活泼氢的完全交换,但是这种方法成本极高;也有通过改变进样方式等方法实现交换,但是这些方法都存在着交换率不高的缺点。
因此,提出一种经济有效的交换方法是使 H/D 交换技术得以广泛普及的动力,同时,目前已有研究者通过简单的柱后补偿D2O结合计算的方法实现了对 H/D 交换程度的描绘,并成功计算出痕量杂质中活泼氢的个数。
结构解析时,对质谱的每个数据给出合理的解释是十分重要的,同时其他数据对质谱解析也非常有帮助,例如HRMS的分子离子峰和同位素峰计算出的分子式可从模拟合成杂质数据库找出可能的结构,再以 H/D 交换的数据验证结构准确程度;对降解产物研究时,使用多级碎片离子质量、丢失的中性分子质量以及活泼氢数据与药物活性成分的相关数据进行对比的方法可快速准确判定杂质的结构。
2.4.2 核磁共振技术(NMR)
NMR 技术在杂质定性和定量应用中主要依赖于获得杂质单体,另外,在特殊杂质质量标准建立时对杂质对照品的标化可利用定量NMR进 。NMR技术同时也是一种质量相关检测技术,使用NMR技术求算校正因子进而校正其他检测器,可实现对反应进程的监控。NMR技术检测灵敏度依赖着探针的性能。
因此,为了提高NMR检测灵敏度,有研究者发明了致冷探针,这种探针只需微克级别的化合物单体就能实现对化合物检测,这使NMR检测样品消耗量实现了由毫克向微克的飞跃。同时在线的液相-核磁(LC-NMR)联用技术也能实现药物杂质的快速结构鉴定 。
2.5 新型的分析技术
随着快速杂质分析以及结构鉴定准确性的需要,也有一些方法被用于杂质的结构鉴定,如药物杂质直接测定技术 、以分子印迹法(MIP)建立同类杂质碎片数据库以及单晶 X 线衍射技术等。
3 基因毒性杂质的分析
基因毒性杂质能直接作用于人体中的 DNA,造成 DNA 损伤而具有致癌、致畸或致突变的性质。少量的基因毒性杂质也能对人体造成极大的损害,对基因毒性杂质的研究已引起了药物研究者的广泛关注 。
3.1 定量分析
由于基因毒性杂质危险性极大,美国 FDA 和欧洲药品管理局(EMA)规定了毒理学担忧阈值(TTC):人在长期用药时,潜在毒性杂质每日摄入量不能超过 1.5 μg 。
根据上述规定,每天药物服用剂量是 200mg,则毒性杂质的质量分数不应超过7. 5 ×10-6,因此对基因毒性杂质定量分析需要痕量分析的方法。常用的紫外检测器最低能够对质量分数5 ×10-4的杂质进行检测,但随着 2D-HPLC的发明以及高灵敏度紫外检测器的使用,药物中痕量杂质经一维液相仪分离、固相萃取技术富集及二维液相仪高灵敏度检测也能实现准确定量。
但是,这种方法需要多次高浓度进样并不适合杂质含量测定。
因此,质谱检测技术是建立基因毒性杂质定量分析方法的重要前提。Kakasaheb等 对坎地沙坦中合成起始原料中的基因毒性杂质利用 GC-MS 的方法进行定量测定,方法学验证结果能够符合 ICH 规定的要求,该方法适用于上市药品中基因毒性杂质的含量测定。
在痕量的杂质进行方法建立过程中,样品的前处理方式十分重要,微量的损失可能对结果造成极大的影响,Devenport 等创新性地在大气压条件下利用质谱直接定量出模拟药物中基因毒性杂质,这种方法使样品测定更加便捷,并可实现对药物的高通量检测。
3.2 毒性杂质的确定
2006 年,EMA 发布了遗传毒性限度的指导原则,此后 ICH、FDA 以及我国的药品审评中心也提出了对基因毒性杂质的研究和控制方案。根据相关的指导原则,即使杂质的含量在规定的定量要求以下也需要对杂质进行毒性评估。
对化合物毒性评估是通过体外细菌毒性实验实现的,同时指导原则推荐使用杂质单体进行研究。然而,药物中的毒性杂质都是微量或痕量存在的,通过制备等方法获取杂质单体耗时长且成本高。
为此,FDA 和其他公司共同开发了计算机软件用于评估化合物的毒性。此软件无需获取单体,对杂质的结构进行鉴定后,即可使用毒性评估软件评估杂质的结构是否存在 警 示 结 构。
在软件方面,FDA 推荐使用MC4PC、MDL-QSAR 以及 Derk for Window 等进行评估。为了对毒性得以准确性的评估,每种软件拥有特殊的算法和适用范围:
MC4PC软件是将待评估的结构拆分成 2 ~3 个(非氢)原子的结构碎片,再将这些碎片与已知毒性化合物碎片数据库进行比对,对杂质结构中的碎片做出了毒性累加得分并描绘结构的分子特征,最终生成一个全面而且专业的报告评估杂质的结构毒性。
虽然 MC4PC能够从杂质的精细结构角度评估药物的毒性,但是其只能在已有的数据库中搜索相同的碎片进行比较,对多于一种未知碎片的结构却不能全面评估。
毒性化合物的研究结果表明毒性化合物结构中都存在着亲电子基团或可被激活为亲电子基团的结构,MDL-QSAR毒性评估软件是基于化合物的毒性和该化合物的亲电能力的相关性做出评估,这种软件可计算出化合物结构与其毒性的定量关系,同时可以预测体外细菌毒性实验的结果。
然而,为了达到对已知结构的杂质毒性的最准确评估,使用 MDL-QSAR软件时需寻找到最适合的模型用以计算,因此在该计算软件提出后,有许多关于不同的数据模型以及对它们的评估的报道。
有些杂质虽经计算机评估结果为有警示结构的潜在毒性杂质,但由于评估软件在计算过程中会过分评估杂质的毒性,所以在判定药物杂质是否为毒性杂质仍需要进行体外细菌回复突变实验(Ames 实验)或哺乳动物细胞分析,FDA对该实验的过程给予详细的说明。
3.3 控制方法
根据 EMA 指导原则以及对指导原则问题解答,药物中所有存在的化合物均应该通过化合物毒性评估或符合指导原则的控制方法,同时为了确定潜在毒性杂质是否为毒性杂质需要进行 Ames实验。
4.展望
杂质研究在药物研发过程中占有及其重要的地位,不仅影响着新药上市的速度,更影人类用药安全。随着分析技术的不断发展,杂质研究的策略已经变得愈发多样化。随着检测器分辨率以及灵敏度的提高,杂质定量实现了由微量到痕量的飞跃,杂质的定性工作也实现了由离线到在线检测的转变。然而,新药研发速度的加快,生物药物的不断涌现以及快速定性定量分析的要求,将给研究人员带来极大的挑战。

来源:中国药科大学学报


