在液体制剂研发中,不可避免的问题是滤芯的选择,冻干粉针通过滤芯过滤除菌,大输液和小水针通过滤芯过滤减菌,口服溶液通过滤芯过滤控制可见异物等。如果选择合适的滤芯?
依据最新版药品GMP相关法律和法规、欧盟GMP指南、美国FDA《无菌工艺指南》及《除菌过滤技术及应用指南》等相关法规和指南的要求,选用的滤芯需要开展滤芯相容性研究,滤芯与药液相容性良好才可以在生产中使用。滤芯相容性研究包括哪些呢?
本文关于滤芯相容性研究,进行简单的介绍。滤芯相容性研究一般包括吸附试验、细菌截留试验、化学兼容性试验、可提取物或浸出物试验、安全性评估等内容。
研发工作者需要考虑滤芯相容性的情况,结合研发品种,选择合适的滤芯。
本文结合滤芯相容性研究的5大板块,给出滤芯选择的策略。
一、吸附试验
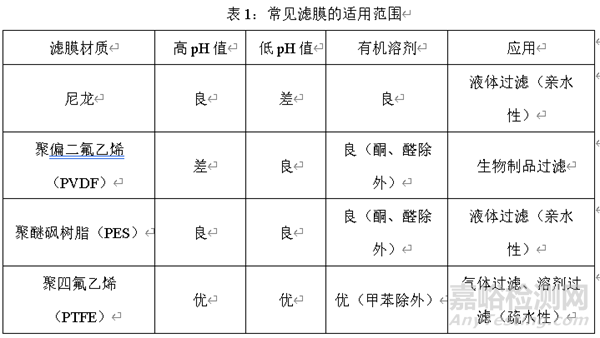
1、在滤芯选择时,首先要充分考虑滤膜的适用范围,包括过滤溶液的pH值范围,过滤溶液的组成成分,过滤器应用的场景等。
首先考虑药液的pH值范围。
例如,过滤的药液pH值偏高,不适宜选择聚偏二氟乙烯的滤膜,推荐使用聚四氟乙烯滤膜。而过滤的药液pH值偏低,不宜选择尼龙滤膜,推荐使用聚四氟乙烯滤膜。也就是说,不论是在酸性环境还是碱性环境均适宜聚四氟乙烯滤膜。
其次考虑药液的组成。过滤溶液组成成分不同,选择的滤膜也不同。含过滤溶液组成中有机相,推荐使用聚四氟乙烯滤膜。
最后,在应用场景方面,尼龙和聚醚砜树脂的过滤器适用于液体过滤,聚四氟乙烯为疏水性滤膜适用于气体过滤,因为改滤膜具有疏水性,进行溶剂过滤时,需要充分润湿。
下表将常见的滤膜适用范围进行梳理,研发工作者可以结合开发的品种性质对照下表进行选择:
2、滤芯选择要考虑滤芯材质与原料、辅料的相互作用,充分考虑过滤器与药液的兼容性。
案例一:滤芯与原料相互作用
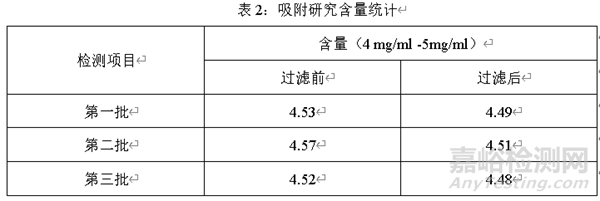
一般过滤器在过滤药液过程中,或多或少会吸附原料。在废弃一定药液后,过滤前后对主药含量进行检测,考察药液废弃不同体积后的含量变化,以确定滤芯对原料的吸附影响。
选择三个批次药液,分别在过滤前和过滤后测定含量,对比过滤前和过滤后含量的变化,结果见下表
经过三批过滤药液前和过滤药液后的含量可知,过滤后的药液含量与过滤前药液的含量相比略微降低,但是过滤前后含量无显著变化,所以,该过滤器对过滤药液无吸附。
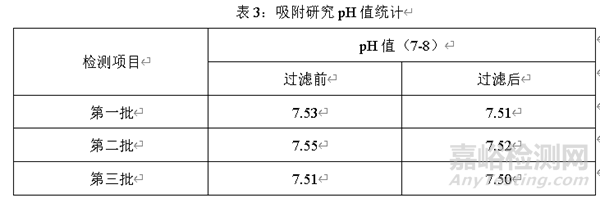
除了考虑药液过滤前后含量的变化,还应考虑过滤前后药液pH值变化。选择三个批次药液,分别在过滤前和过滤后测定pH值,对比过滤前和过滤后pH值的变化,结果如下
经过三批过滤药液前和过滤药液后的pH值可知,过滤后的药液pH值与过滤前药液的pH值相比略微降低,但是过滤前后pH值无显著变化,通过pH值变化情况,进一步证实该过滤器对过滤药液无吸附。
生产过程中出现含量偏低的几个考虑
如果过滤的药液API含量低,滤芯吸附的影响会相当显著。例如激素类药物,API在处方组成中占比小,滤芯的吸附对API含量的影响极大,在药液正式灌装前,要充分考察药液废弃体积。另外,滤芯对API的吸附与滤芯的尺寸成正相关,滤芯的尺寸越大,滤芯吸附API达到饱和的量越大;滤芯的尺寸越小,滤芯吸附API达到饱和的量越小。针对API在处方组成中占比小的情况,可以选择5英寸或者定制更小尺寸的滤芯。
另外,在生产过程中,要充分考虑滤器在线灭菌的影响,滤芯通过在线灭菌后,会有部分水残留在滤器中,即使通过压缩空气或者氮气吹扫,仍然不能把滤器的残留水全部吹出。这部分残留水需要用药液进行充分的置换才能排出。
生产管路在进行清洁和蒸汽灭菌后,管路降温,部分蒸汽会冷凝附着在管路内壁形成残留余水,如果车间管路设计不合理,部分残留余水无法通过排水口排出,需要通过压缩空气或者氮气进行吹扫,吹扫的时间和压力不同,残留余水的量也不同。需要控制管路吹扫时间和压力,确保滤芯和管路的残留余水在可接受范围内。
案例二:滤芯与辅料相互作用
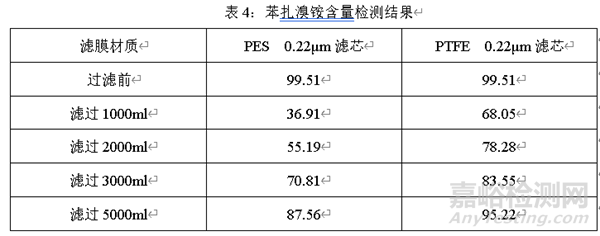
在研发过程中,要考虑处方的某些成分与滤器的相容性。过滤器在过滤药液过程中,可能会吸附某些辅料,例如苯扎氯铵、苯扎溴铵。在过滤前检测苯扎溴铵含量,过滤不同体积,检测过滤不同体积后苯扎溴铵的含量。
经过上述考察可知,不同材质滤芯对苯扎溴铵均有吸附。在废弃相同体积后,PTFE过滤的药液苯扎溴铵含量比PES更高,说明PTFE对苯扎溴铵的吸附与PES相比更小。
在处方开发过程中,要充分考虑特殊辅料(如:苯扎溴铵、苯扎氯铵等)与滤芯的相容性。
除了吸附的影响,过滤器不得因与产品发生反应、释放物质而对产品质量产生不利影响。
总之,在滤芯吸附试验板块,滤芯的选择,首先要考虑滤芯的适用范围,其次是滤芯与原料、辅料以及药液的相容性。
二、影响细菌截留的因素
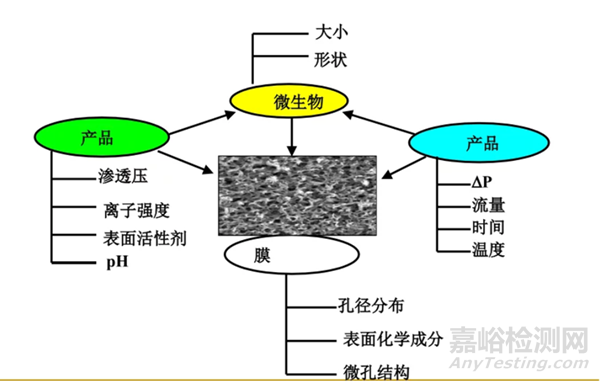
首先是微生物的性状和大小;
其次是药液的性质,包括渗透压、离子强度、pH值、表面活性剂占比、有机成分占比、抑菌或杀菌成分的占比等;
然后是滤膜的性质,包括孔径分布、表面化学成分。微孔结构等;
最后是药液通过滤膜的压差、时间、温度、批量等。
影响细菌截留的因素详见下图:
图5:影响细菌截留的因素
三、细菌截留的研究方式
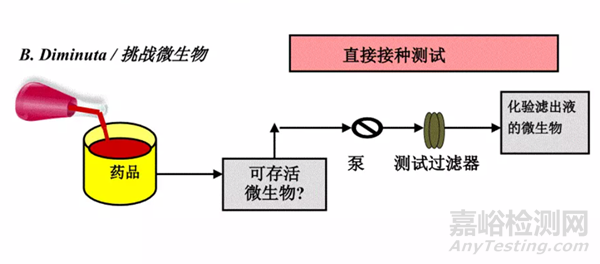
细菌截留的研究方式一般有两种:
直接法和间接法。
在开展细菌截留研究前,要先确认药液对细菌的影响。确认药液对细菌的影响分为4步:
第一步
在生产工艺条件下,将挑战菌直接加入到产品中,并在工艺条件下进行培养。
第二步
检测产品中挑战菌的存活率。
第三步
根据挑战菌在测试液和对照液中的LRV值不同,可以将测试液分成杀菌性产品、非杀菌性产品和中度杀菌性产品三种。
第四步
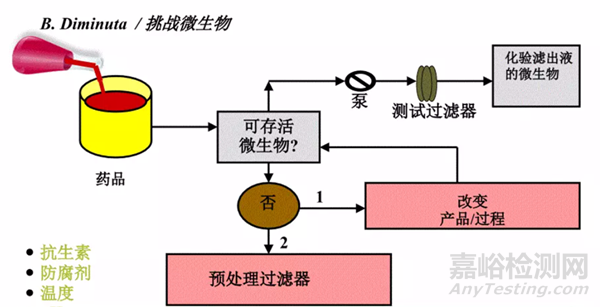
如果产品为非杀菌性产品,则细菌挑战测试的方式为直接将挑战菌加入产品中进行挑战;
如产品为中度杀菌性产品或杀菌性产品,则需要建立滤芯或滤膜的冲洗方案,验证产品残留对细菌生存性的影响,并选择合适的替代液代替客户产品进行细菌挑战测试。
图2:细菌截留研究方式——直接法
图3:细菌截留研究方式——间接法
判断标准
1、在工艺时间内活菌数下降<1log,确定测试药液为非杀菌性产品。
2、1h内活菌数下降<1log,但是在工艺时间内活菌数下降≥1log,确定测试药液为中度杀菌性产品。
3、1h内活菌数下降>1log,确定测试药液为杀菌性产品。
4、恢复生长的菌落数必须在 20~200 cfu 才可作为有效计数结果。
四、细菌截留试验需要考虑的方面
1、在除菌过滤验证中使用滤膜还是滤器,取决于验证的目的。如果微生物截留试验的目的是验证过滤工艺中特定膜材的细菌截留效能,那么使用滤膜是能满足需要的。微生物截留试验中所用的滤膜必须和实际生产中所用过滤器材质完全相同,并应包括多个批次(通常三个批次)。其中至少应有一个批次为低起泡点(低规格)滤膜。
2、为了在微生物挑战试验中实施最差条件,一般需要使用完整性测试的数值非常接近过滤器生产商提供的滤器完整性限值的滤膜(例如不高于标准完整性限值的110%) 。如果在验证中没有使用低起泡点滤膜,那么在实际生产中所使用的标准溶液滤膜/芯起泡点值,必须高于验证试验中实际使用的滤膜的最小起泡点值。
3、成分相容、只有浓度不同的系列产品可以通过挑战极限浓度和接受中间一组浓度来验证。
4、一般来说重复使用过滤器是不实际的,如果重复使用除菌级过滤器,应该给出充足里有并且重复使用参数经过充分论证。
结合细菌截留试验的研究内容,关于滤膜孔径选择和滤芯组合系统选择,本文推荐如下:
关于滤膜孔径的选择,在这里,要先科普一下,世界上最小的细菌是缺陷型假单胞菌(直径大约为0.3—0.4微米,长度0.6—1.0微米)。在除菌过滤验证中,缺陷型假单胞菌必须是单一的、分散的细胞,才能作为细菌截留试验的标准挑战微生物。所以只有过滤器孔径小于缺陷型假单胞菌的尺寸时,才能保证对最小微生物的截留能力。一般情况,除菌过滤工艺应选用0.22微米(更小孔径或相同过滤效力)的除菌级过滤器。
滤芯组合一般分为两种:序列过滤系统和冗余过滤系统。
通常通过两个或以上相同或递减孔径的过滤方式,统称为序列过滤系统。例如:0.45μm过滤器+0.22μm过滤器。序列过滤系统中,如果在最终除菌过滤器前增加一个除菌级过滤器,并且确保两个过滤器之间无菌,以及控制过滤前介质的微生物污染水平一般小于等于10cfu/100ml,这种情况下称为冗余过滤系统。例如:0.45μm过滤器+0.22μm过滤器+0.22μm过滤器。
总之,通过细菌截留试验研究和论证,除菌过滤工艺应选用0.22微米(更小孔径或相同过滤效力)的除菌级过滤器,除菌过滤工艺一般选择冗余过滤系统,达到更高的无菌保障水平。
五、化学兼容性试验
根据国内外GMP相关条款要求:与药品直接接触的滤器不得与药液发生化学反应、吸附药品或者向药液中释放物质。
化学兼容性试验要包括滤芯的全部组成部件(包括滤膜,滤壳外骨架、端盖支撑部件,密封圈等)。
在最差实际过滤工艺条件下,考察对滤芯过滤性能的影响,包括2个方面:
1是药液是否影响滤芯过滤性能,
2是滤芯是否影响药液性质。
过滤工艺要考虑待过滤介质性质、过滤温度、接触时间、过滤批量、过滤批次等。考虑过滤介质性质,例如过滤介质是否含有乙醇等有机相,有机相的组成比例,试验过程中的过滤温度应达到或者超过生产过程的最高温度,过滤时间应达到或者超过实际生产过程的最长工艺时间。
化学兼容性试验检测项目一般包括:
1.滤芯
过滤器接触待过滤介质前后的目视检查,要求外观无损坏、变形和软化现象,起泡点值大于或者等于3200mbar(测试液体为水),扩散流值小于等于10ml/min/5in@0.28Mpa(测试液体为水),浸泡前后相对偏差不超过10%。
2.支撑部门
过滤器接触待过滤介质前后目视检查,要求外观无破损和软化现象。
3.滤膜
接触过滤介质前和接触过滤介质后,要求外观无破损和软化现象,过滤过程中流速变化,一般要求不超过20%;滤膜重量是否变化,重量变化一般不超过5%;厚度是否变化,厚度变化一般不超过5%;滤膜拉伸强度的变化,拉伸变形率一般不超过5%;滤膜电镜扫描确认无明显差异。
4.密封圈
接触过滤介质前和接触过滤介质后,要求外观无变形、溶胀和软化现象,密封圈内径变化要求浸泡前后相对偏差在±2%之内,重量要求浸泡前后相对偏差在±2%之内。
5.药液
接触过滤介质前和接触过滤介质后,要求无肉眼可见的滤芯脱落物。
特别需要注意的是,滤器接触药液后,药液中可能存在部分颗粒堵塞滤膜孔,进而导致浸泡后滤芯的泡点和过滤阻力增加,所以,过滤后起泡点相对偏差不设置上限。
在化学兼容性部分,要考虑滤芯的过滤阻力大小、滤膜厚度、拉伸强度,结合车间生产线的实际情况,选择筒式或者囊式滤芯。
六、可提取物或浸出物试验
根据NMPA的GMP(2010版)提出了如下要求:生产设备不得对药品质量产生任何不利影响。因此要关注生产设备(例如过滤器)与工艺溶液接触会后,其结构可能发生改变,进而导致过滤器组分迁移至药品中。
可提取物是在极端条件下(例如有机溶剂、极端高温、pH值、接触时间),可以从过滤器提取出的化学物质。浸出物是在正常工艺条件和试机药品中,从过滤器迁移出来的化合物。
使用最长过滤时间、最高过滤温度、最多次蒸汽灭菌循环、增加伽玛辐射的次数和剂量都可能会增加可提取物水平。
可提取物反映了浸出物的最大可能。
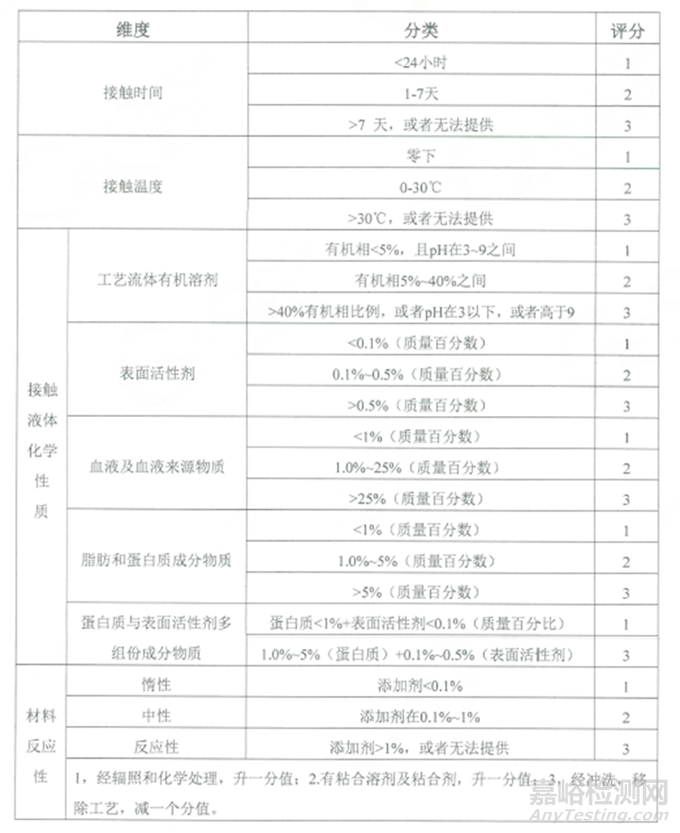
可提取物试验可以用静态浸泡或循环流动的方法,其影响因素包括接触时长、接触温度、溶液类型和组件材料活性。
表1:可提取物维度、分类及评分汇总表
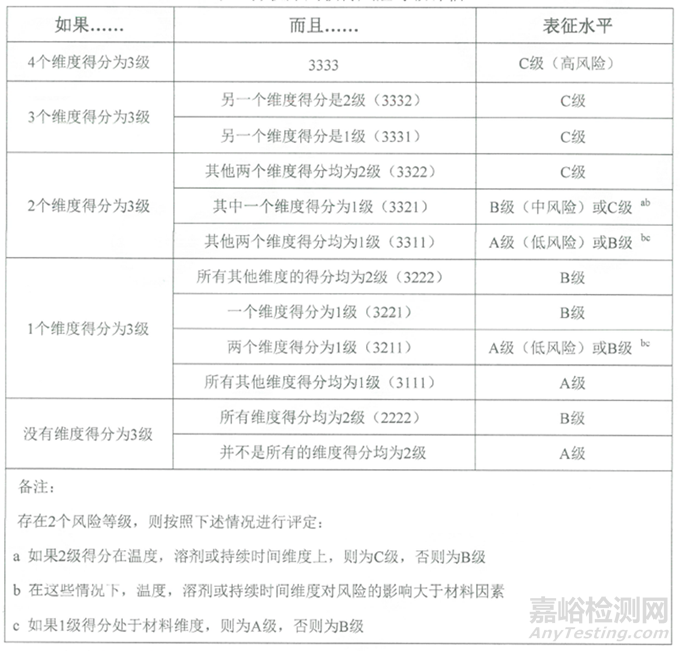
根据接触时长、接触温度、溶液类型和组件材料活性等维度的评分,评估出提取物研究的风险等级。提取物研究的风险等级详见下表:
表2:提取物研究的风险等级评估表
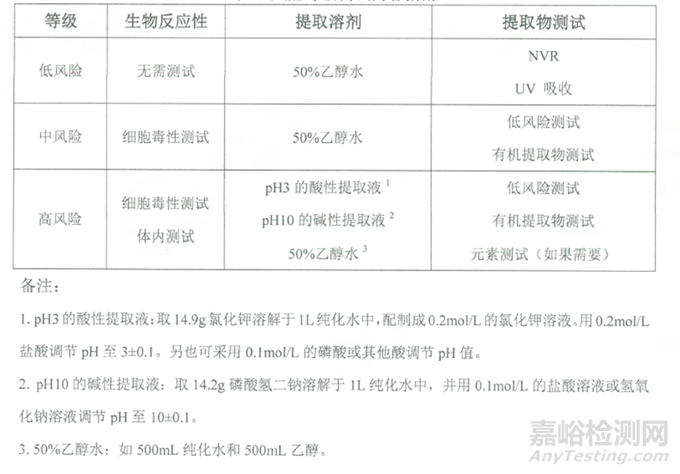
根据提取物评估的风险等级,开展相应的研究,不同等级对应的提取物测试汇总表如下:
表3:不同等级对应的提取物测试汇总表
提取物或浸出物研究包括:
1、 不挥发物(NVR)检测可提取物中非挥发物
2、 紫外可见吸收光谱(UV-VIS)检测发色物质的官能团
3、 傅里叶变换红外光谱法(FTIR)检测聚合物或寡聚物的官能团
4、 反相高效液相色谱(RP-HPLC)检测溶剂、单体和添加剂
5、 气相色谱-质谱(GC-MS)挥发及半挥发物
用于试验的过滤器尽量不进行预冲洗。在可提取物和浸出物试验部分,要结合处方组成(有机相比例),工艺参数(过滤温度、过滤时间、过滤体积和批量),灭菌方式(在线灭菌还是离线灭菌)等因素,选择适宜的滤芯。
七、安全性评估
在完成可提取物或者浸出物试验后,应针对过滤器可提取物或浸出物的种类和含量,结合药品最终剂型中的浓度、剂量大小、给药时间、给药途径等对结果进行安全性评估,以评估可提取物和浸出物是否存在安全性风险。
参考文献
1.国家食品药品监督管理局.药品生产质量管理规范(2010年修订).2011.
2.US Food and Drug Administration, Current Good Manufacturing Practice for Finished Pharmaceuticals, Equipment Construction, 21 CFR Part 211.65(a), 2019.
3.European Commission, EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Premise and Equipment, Chapter 3.39, 2015.
4.U.S. Department of Health and Human Services, Food and Drug Administration, Guidance for Industry, Container Closure Systems for Packaging Human Drugs and Biologics, May 1999.
5.Parenteral Drug Association (PDA), Sterilizing filtration of liquids technical report No.26, 2008
6.国家食品药品监督管理局药品认证管理中心.无菌药品:药品GMP指南. 2011.
7.国家药品监督管理局.除菌过滤技术及应用指南(2018年第85号通告) .2018-07-31.
8.国家药品监督管理局药品审评中心.化学药品注射剂生产所用的塑料组件系统相容性研究技术指南(试行)(2020年第33号通告).2020-10-21
9.Ding, W., et al., "Standardized Extractables Testing Protocol for Single-Use Systems in Biomanufacturing," Pharmaceutical Engineering, Vol. 34, No.4, 2014.
10 United States Pharmacopeial Convention, Plastic Components and Systems Used to Manufacture Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Products, <665>,2022.
1.American Society for Testing and Material. Standard Test Method for Determining Bacterial of Membrane Filters Utilized for Liquid Filtration. 1993 ASTM Standard and Environmental Microbiology,Second Edition, Designation F838-83, pp 50-55.
2.Center for Drugs and Biologics and Office of Regulatory Affairs, Food and Drug Administration. Guideline on Sterile Drug Products Produced by Aseptic Processing. September2004.
3.PDA Journal of Pharmaceutical Science and Technology: Technical Report No. 26, Sterilizing Filtration of Liquids, March 1998.
4.国家食品药品监督管理局,药品生产质量管理规范(2010年修订) [S].2011
5.国家食品药品监督管理局药品认证管理中心,无菌药品:药品GMP指南[S].2011
6.国家药品监督管理局.除菌过滤技术及应用指南(2018年第85号通告) [2].2018-07-31
7.国家药品监督管理局,除菌过滤技术及应用指南,2018
8.Parenteral Drug Association(PDA), Sterilizing filtration of liquids technical report No.26,2008
9.General Information Chapter<1227>,Validation of Microbial Recovery from PhamacopeialArticles, USP 28,USPC,Inc.,Rockville,MD,2005,p. 2752
10.American Society for Testing and Materials (ASTM) Standard Test Method for Determining Bacterial Retention of Membrane Filters Utilized for Liquid Filtration F838-05,pp1-6,ASTM,WestConshohocken,PA,2005.
1.国家食品药品监督管理局.药品生产质量管理规范(2010年修订).2011.
2.US Food and Drug Administration, Current Good Manufacturing Practice for Finished Pharmaceuticals, Equipment Construction, 21 CFR Part 211.65(a), 2019.
3.European Commission, EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Premise and Equipment, Chapter 3.39, 2015.
4.U.S. Department of Health and Human Services, Food and Drug Administration, Guidance for Industry, Container Closure Systems for Packaging Human Drugs and Biologics, May 1999.
5.Parenteral Drug Association (PDA), Sterilizing filtration of liquids technical report No.26, 2008
6.国家食品药品监督管理局药品认证管理中心.无菌药品:药品GMP指南. 2011.
7.国家药品监督管理局.除菌过滤技术及应用指南(2018年第85号通告) .2018-07-31.
8.国家药品监督管理局药品审评中心.化学药品注射剂生产所用的塑料组件系统相容性研究技术指南(试行)(2020年第33号通告).2020-10-21
9.Ding, W., et al., "Standardized Extractables Testing Protocol for Single-Use Systems in Biomanufacturing," Pharmaceutical Engineering, Vol. 34, No.4, 2014.
10 United States Pharmacopeial Convention, Plastic Components and Systems Used to Manufacture Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Products, <665>,2022.
11. Container Closure Systems for Packaging Human Drugs and Biologics
U.S. Depaitment of Health and Human Services Food and Drug Administration Center for Drug
Evaluation and Research (CDER) & Center for Biologics Evaluation and Research (CBER). May
1999
12. 国家食品药品监督管理总局,化学药品与弹性体密封件相容性研究技术指导原则,2016
13. GB/T 34244-2017 液体除菌用过滤芯技术要求