1、背景
生物类似药,是指在质量、安全性和有效性方面与已获准上市的参照药具有相似性的治疗性生物制品[1]。其中的相似性,是指候选药与已获准注册的参照药整体相似,且在质量、安全性及有效性方面不存在有临床意义的差别[2]。这部分的定义,在中美法规指南中基本一致。是否存在临床意义的差别,是相似性评价的考察点。
早在2015年,美国FDA的两份指导原则[3][4]和中国国家药监局的指导原则[1]提出,可采用递进式的方式证明候选药与参照药的相似性,并且提示需要在药学质量相似性研究中考虑各质量属性与临床的相关性,甚至应根据各质量特性与临床效果相关的程度确定评判相似性的权重,并设定标准。
2017年,美国FDA发布指导原则草案[5],提出应采用风险评估工具,评估产品质量属性对临床的潜在影响,并将质量属性分为三个级别,临床相关性最强风险最高的质量属性为第一级(Tier 1,通常是生物学活性方法),风险更低的为第二级(Tier 2),风险最低的为第三级(Tier 3),并且给出了每个级别所推荐使用的统计学分析工具,Tier 1使用等效性检验(Equivalence testing),Tier 2使用质量范围(Quality ranges)评估,Tier 3可采用直观对比(Visual comparisons)的方式。虽然该指导原则草案在公示不到一年的时间就被FDA撤回,不过撤回的原因主要是业界反馈认为,指南中要求参照药及生物类似药的批次数量、统计学的评估方法影响了生物类似药开发的成本和效率,而其中提出的基于临床影响的质量属性分级思想依然被业界所广泛接受。
2019年,FDA发布指导原则草案[6],重新提出了对生物类似药质量相似性数据分析的建议,认为对于高到中等风险的质量属性,可以使用质量范围的方法进行评估,对于低风险或无法定量的质量属性采用直观对比的方式进行。同时提到,不推荐使用容忍区间作为相似性的接受标准(因为获得有意义的区间需要较大的样本量),但可以使用其他统计方法,如等效性检验。
2021年,中国国家药监局发布了《生物类似药相似性评价和适应症外推技术指导原则》,在之前指导原则的基础上,明确了应“基于对参照药质量属性的认知程度及其与临床风险获益的相关性,采用合适的风险评估工具对质量属性进行分级”,并且,“质量相似性定量评估引入统计学分析方法有助于增强相似性判断的客观性”,推荐使用质量范围(μR-XσR, μR+XσR)和直观对比的方式进行,“对于和产品作用机制或临床表现直接相关的质量属性(如生物学活性),可根据质量风险分级和相似性评价目标,采用其他统计方法进行数据分析,如等效性检验。”
时至今日,在实际操作中,药学部分的质量相似性评价依然有一些矛盾和挑战,大家往往会被诸如参照药不同原液批次的收集、代表性样品的批次数量、实施中的统计工具、判断相似的可接受标准等等问题困扰。本文即围绕统计工具和可接受标准这一点进行探讨,以加深对工具的理解,帮助具体的应用。这些统计工具不仅应用于质量相似性评价中,而且能够广泛地应用在变更可比性研究、技术转移、分析方法对比、维护乃至样品检测等各个方面。
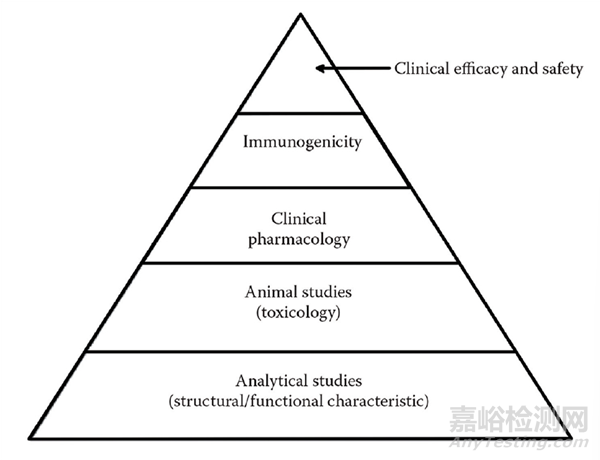
图 1 Stepwise approach to demonstrating biosimilarity
2、风险评估和分级
质量相似性评价指标通常包括蛋白结构和理化特性、纯度和杂质、生物学活性、免疫学特性、稳定性等[2]。质量属性的风险评估可以采用多种风险评估工具进行(如RPN风险优先顺序数评估、RRA风险排序分析法等等),但至少需要包含如下两个方面[6]:
对临床表现的潜在影响:包括活性、PK/PD、安全性、有效性和免疫原性,可以通过公开可获得的信息,或自行对参照药进行的相关表征研究获得信息。
围绕某一质量属性的不确定程度:如质量属性与临床表现之间的相关性存在不确定性时,其质量风险应评估为更高的等级。
稳定性比对研究(例如稳定性或强制降解研究中的降解速率等)可不必进行风险分级,但应体现在质量相似性评价中。
3、Tier 1 (Equivalence testing) 等效性检验
对于和产品作用机制或临床表现直接相关的质量属性(如生物学活性),被分类为Tier 1,推荐使用等效性检验方法进行评估(也可使用质量范围方式进行评估)。FDA撤回指南[5]的统计学评估方面的原因也主要在等效性检验方面[7]。
自2014年起,FDA就开始通过PINDs和INDs的会议意见向申办方推荐等效性检验的方法[7]。从技术角度而言,等效性检验是目前用来判断二者是否等效的首选方法。我们常用差异性检验(如t检验)来评估两组数据的均值差异,但是当两组数据的均值差异没有统计显著性时(如p值大于0.05),我们只能得到两组数据差异不显著的结论,而无法得到两组数据等效的结论。等效性检验能够很好地解决这个问题。此外,等效性检验不仅计算了差异的大小,还考虑了差异的意义,从而得到等效与否的结论。
简单地说,等效性检验是一种假设检验,它通过假设参照药质量属性的均值μR,与候选药质量属性的均值μT,二者之间的差异μT-μR,应在一个能够被接受的范围内(或者说在一个被认为是没有临床影响的范围内),即等效界值δ,其假设为:
● H0: μT-μR ≤ -δ or μT-μR ≥ δ
● Ha: -δ < μT-μR < δ
该假设也可以写成一种等价形式:
● H01: μT-μR ≤ -δ vs. Ha1: μT-μR > -δ
● H02: μT-μR ≥ δ vs. Ha2: μT-μR < δ
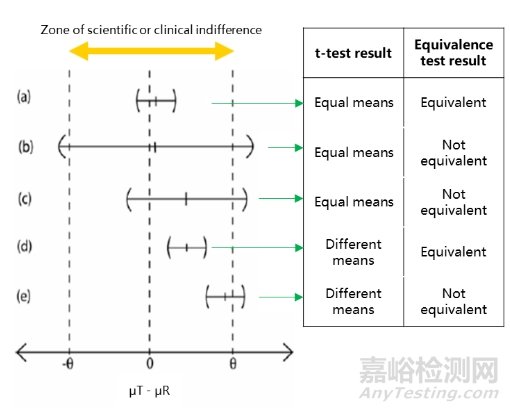
因此,等效性检验可以通过两种方式进行,一种是计算μT-μR差值的90%置信区间(α=0.05),当等效界值完全包含置信区间时,等效性成立,另一种是进行双单边t检验(TOST),在-δ和δ两侧分别进行单边t检验(α=0.05),当两个p值均小于0.05时,说明等效性成立。两种方法完全等价。
图2 左:等效性检验将差值的90%置信区间与等效界值进行对比;右:t 检验与等效性检验的结果对比
大部分统计分析软件都具备计算等效性检验相关统计量的功能,甚至使用Excel或者手动即可进行相关统计量的计算。统计计算本身不是很大的问题,最大的问题通常在于等效界值的设置(即可接受标准的设置)和样本量的确定。
等效界值的设置对等效性检验来说是最困难且最关键的环节。理想情况下,可以基于科学知识或过往经验预设一个生物学意义或临床意义的等效界值,但受到药物研发阶段等的限制,通常无法实现。因此,FDA建议基于参照药标准偏差σR,采用f×σR作为等效界值,在建议批次数量n ≥ 10的条件下,为满足检验水准(α=0.05)和检验效能(power ≥ 85%),FDA认为f取值为1.5是比较合适的[5],即δ = 1.5×σR。
值得注意的是,FDA的以上建议,是基于以下假设进行的:
假设均值的真实差异与σR成正比,即μT-μR与σR成正比;
σR通过参照药样品检测值的标准偏差获得(一个批次有一个检测结果);
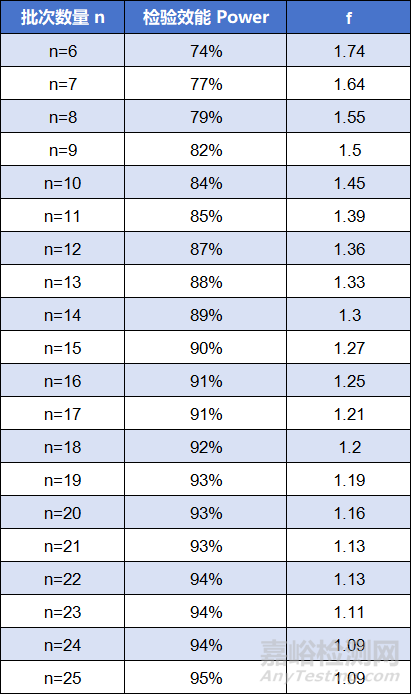
为获得期望的等效性检验的检验效能,FDA进一步建议在μT-μR = 1/8σR的假设下,计算样本量。
基于均值的真实差异为1/8σR和检验水准α=0.05的前提下,批次数量、检验效能及f的取值有如下关系[8]:
然而,上述假设引起了业界和学术界的大量讨论,其结论是值得商榷的。
在使用该种方法时,由于参照药质量属性的总体标准差σR未知,需要从已有样品的检测中获得(即用样品标准差s替代总体标准差σ),而参照药的产品批次来源在本质上是未知的,参照药批次间的相关性(如不同参照药批次可能来自同一原液批次)直接影响了相似性评价的设计和评估[7]。也有研究指出[9][10],在进行等效性检验时,通过计算样品标准差s作为总体标准差σR,并在假设检验中视之为常数,以此在正态分布假设中进行统计量的计算,会导致第一类错误(Type I error,即等效性检验中结果符合可接受标准,但实际上不符合)的概率膨胀,并使检验效能(power)下降[11]。
为了降低这种不良影响,一种替代的方式是将σR作为参数,然后使用Wald检验,将Wald统计量应用于等效性检验的假设中[5][11]。在这种方法下,为满足检验效能大于85%,在产品批次数量n=10并假设产品变异性相等时,f的取值应从1.5提高至1.7(即 δ = 1.7×σR)。
另外,参照药和候选药批次数量的不均等,也会提高第一类错误的概率并影响检验效能。对此,除了推荐批次数量不小于10以外,通常也建议参照药和候选药的批次数量比例在2/3至3/2之间[12]。当批次数量的不均等非常明显的时候,可采取适当的方法进行调整[13]。在满足检验水准、检验效能的前提下,上述计算假定σT ≈ σR,实际评估中,对于固定的候选药批次数量,当参照药的变异性大于等于候选药的变异性时,参照药的最小批次数量可以小于等于候选药批次数量,而当候选药的变异性大于等于参照药的变异性时,参照药的最小批次数量则需要大于等于候选药批次数量,在这种情况下,甚至存在参照药批次数量无法满足2/3至3/2候选药批次数量的可能。
而且,通常来说,被归类为Tier 1的关键质量属性一般会少于4个。当Tier 1质量属性只有一个的时候,相关的样本量和检验效能计算可按上述讨论进行,而当有不止一个Tier 1质量属性的时候,这些质量属性之间通常是具有相关性的,那么,通过多个等效性检验的检验效能计算会更为复杂[13]。检验效能的计算,同时也与样本量的计算有关,因为等效性检验的参照药样本量 k,会受到以下几个参数的影响:1. 检验的总I类错误率(α);2. II类错误率(β,即等效性检验中结果不符合可接受标准,但实际上符合的概率)或者说检验效能power(1-β);3. 临床或科学有意义的差异(μT-μR);4. 参照药的变异性σR(假定σT ≈ σR)。样本量k即上述参数的函数,k = f(α, β, μT-μR, σR)[14]。对于检验效能,通常设定不低于80%,当进行样本量调整时,建议调整后的检验效能也不低于80%。
基于实际的相似性评价场景,只要能够进行适当的考量和评估,等效性检验的可接受标准设置仍有探讨的空间。
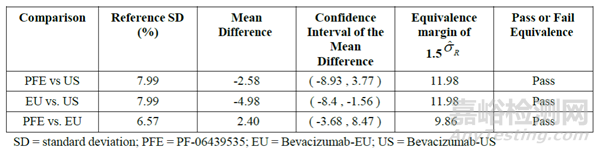
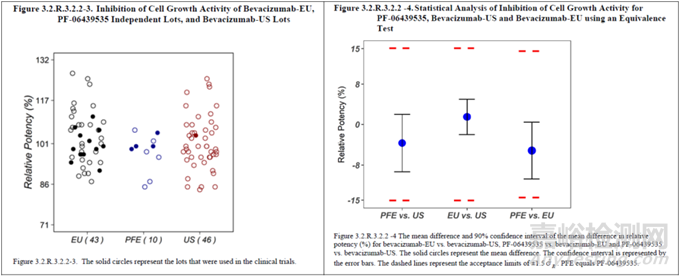
图 3 等效性检验示例(统计量计算表格、散点图及结果的可视化呈现,其中,PFE为拟申报的生物类似药,EU和US分别为不同地区的参照药)[15]
4、Tier 2 (Quality range) 质量范围
对于风险高至中等的质量属性,可采用质量范围的方式进行评估。中美指导原则[2][6]中均有类似描述,质量范围即(μR-XσR, μR+XσR),其中μR为参照药质量属性的平均值,σR为其标准偏差,系数X的设定应根据质量属性的风险等级进行科学论证。只要有足够数量的候选药批次数据(如90%以上)落在质量范围内,即可认为该质量属性与参照药相似(highly similar)。此外,FDA建议,对于风险更高的质量属性,应设置更窄的质量范围进行评估。
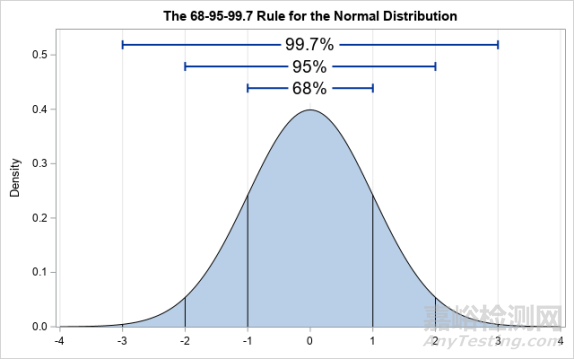
质量范围评估相似性的统计逻辑容易理解,不严谨地说,质量范围即参照药质量属性的分布范围,在正态分布的假设下,当X=3时,质量范围即参照药质量属性的均值±3SD的范围,即大约是参照药质量属性的99.7%的数据所在的范围。在假设μT ≈ μR,σT ≈ σR的前提下,如果候选药的质量属性数据也落在同一个范围内,说明二者很可能服从同一个正态分布,因此可以说明二者的相似性。
我们知道,基于样本数据来推测指定比例的总体所在的统计概率区间,通常使用的是容忍区间的方法。这里仅使用正态分布的范围进行总体数据范围的推测,而未使用容忍区间进行总体数据范围推测,估计也与FDA不推荐容忍区间的原因类似。当数据量较少的时候,容忍区间会得到很宽的接受范围,这个范围也许并不能产生足够的意义,反而增加过高的通过机会(注意,这不代表可比性研究中不能使用容忍区间,需要考虑使用条件)。
对于X的取值,在2015年的杜克大学工业统计研讨会上,一位FDA的发言者指出,X应选择2~3之间的数值[13]。在正态分布的假设下,即代表有95%(当X=2)至99.7%(当X=3)的均值数据落在范围内。
图4 正态分布示例
当然,上述的检验并不完全严谨。首先,我们只能使用有限批次的参照药质量属性检测结果的均值(x-bar)和标准差(s)来代表总体的均值(μ)和标准差(σ),虽然样品标准差(s)是总体标准差(σ)的无偏估计,但确实缺少了对参照药相关变异性的考量,当批次数量有限的时候,这个问题会更为明显。其次,虽然指导原则[2][6]中也提及,需要关注参照药与候选药的平均值和标准偏差的异同,但在实际评估中,我们通常无法确保μT ≈ μR,σT ≈ σR(毕竟我们只能获得 x-bar和s,而非μ和σ),只能基于数据是否满足可接受标准这一结果来判断相似或不相似,当出现μT = μR,σT ≠ σR,或μT ≠ μR,σT = σR,或 μT ≠ μR,σT ≠ σR的情况时,其结果可能会产生误导且变得不可靠。
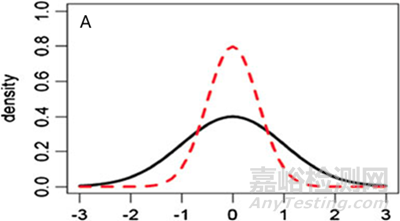
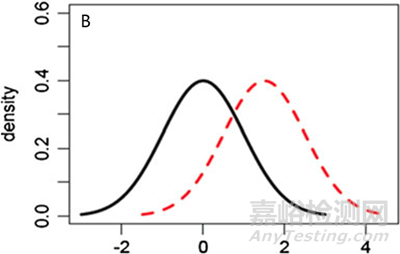
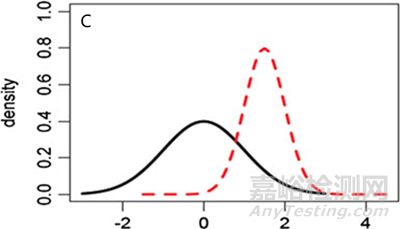
图5 参照药和候选药不同分布情形的图形化示例 (A) μT = μR,σT ≠ σR, (B) μT ≠ μR,σT = σR, (C) μT ≠ μR,σT ≠ σR.
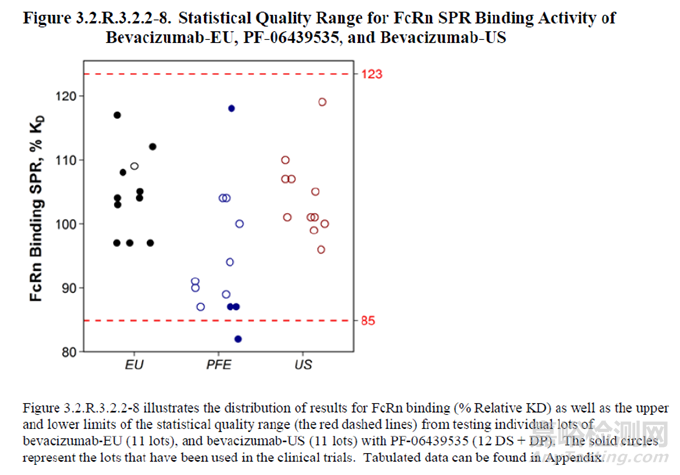
图6 质量范围示例 (mean±3×SD,以散点图形式的可视化呈现,其中,PFE为拟申报的生物类似药,EU和US分别为不同地区的参照药) [15]
5、Tier 3 (Visual comparisons) 直观对比
对于风险低或者无法定量的质量属性,可采用直观对比的方式进行评估。部分质量属性,如蛋白序列、高级结构等,也不适用于定量分析,通常也是用直观对比的方式进行。类似的不适用内容,需要进行清晰的规定,并进行合理化说明。
对Tier 3的质量属性,可以对实际检测数据以恰当的方式(如散点图)进行直观对比。从已公开的各家生物类似药文章和FDA申报资料来看,原始图谱和散点图是被最广泛采用的形式,不仅是在直观对比中,也在等效性检验及质量范围的评估中(这也是FDA指南[6]中推荐的,对于Tier 1和Tier 2的质量属性,也需要进行可视化的展示,以呈现参照药和候选药产品的质量分布)。
原始数据和图像化的呈现,不仅主观,而且可能有偏向性。显然,Tier 1的等效性检验和Tier 2的质量范围评估都比Tier 3的直观对比更为严格。能够通过Tier 1和Tier 2的检验,一定能通过Tier 3的检验。而对于等效性检验和质量范围评估而言,通过Tier 1的检验并不代表能够通过Tier 2的检验,反之亦然[14]。
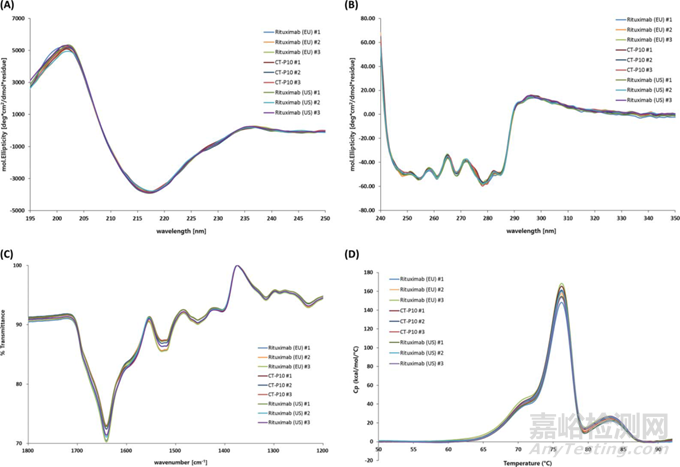
图7 直观对比示例 (通过CD、FT-IR和DSC进行的高级结构分析) [17]
6、分析方法
分析方法是质量相似性评价的基石,只有可靠的检测结果才能支持进行可靠的相似性评价。
指导原则[2]鼓励采用先进的、敏感的、正交的技术和方法对候选药和参照药开展全面的质量比对研究,并尽可能使用不同原理的分析手段检测潜在的质量差异。并且,首选采用与参照药一致的分析方法,采用其他技术和方法的,应评估其优势和局限性,确保其符合检测目的和相似性评价需求。用于放行检测的分析方法应进行全面的方法学验证,用于特性鉴定的分析方法则应确保其可适用于分析目的,分析结果具有可靠性和重现性。
此外,FDA指南[5]中提及,质量属性的变异包含了批内变异和批间变异,需要关注。分析方法的高变异性通常不是设置更大可接受范围的合理化说明,而应该对分析方法进行优化,或提高每批检测时的重复次数,以降低结果的变异性。
7、案例分享
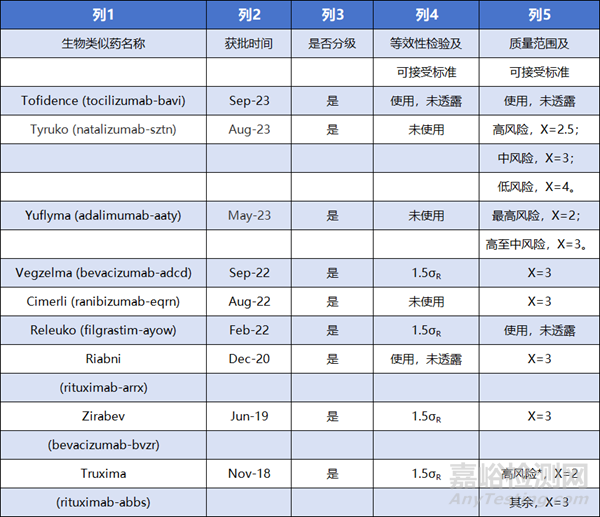
通过查阅FDA官网公开的信息[16],近年来(2018至2023年)获批的生物类似药,其中的质量相似性评价统计分析方法及其可接受标准情况,从中也能推测FDA在质量相似性评价中的审评态度,(表格中仅呈现部分公开了相关信息的药物资料):
*该项目有的质量属性(ADCC)同时采用了等效性检验(1.5σR)和质量范围(X=2)
从资料来看,各项目均基于风险使用了分级的策略进行质量相似性评价。其中,使用了等效性检验的项目中,基本均采用了FDA推荐的1.5σR作为等效界值,当然,作为BLA申报阶段,参照药批次数量通常已经不是很大的问题(Zirabev和Vegzelma项目使用了46批参照药进行评估),这也使等效性检验能够保证足够的检验效能。也有部分项目未使用等效性检验方法,而是采取了质量范围的方法,根据质量属性的风险大小,设置了不同的可接受标准。而这类策略进行合理化说明后,都能被FDA所接受。
结语
从质量相似性评价的统计工具角度出发,回顾质量相似性评价的设计,我们需要关注如下几个方面:
参照药批次的代表性:其批间变异性直接影响了统计分析结果的可靠性;
参照药与候选药的批次数量:为了满足统计分析的检验效能,需要一定的批次数量和比例;在药物研发的早期阶段,因为批次数量不足,部分统计分析方法可能不适用;
质量属性的分级和可接受标准设置:质量属性的分级,以及可接受标准设置的合理性需要基于对数据的理解和对相关影响的评估,统计分析方法本身的局限性也可能影响到可接受标准的设置。
提及统计分析,我们总难免会有一些畏难情绪,部分公司的CMC团队拥有专门的统计团队,可以由专业人士进行相关支持,但需要注意的是,在具体问题的解决中,面对的许多问题并不是统计学家的问题(如活性数据的分布、质量数据的差异和差异背后的意义等),而需要业务部门专家进行判断。数理统计已由统计学家完成,我们的重点是理解并应用这些知识和工具解决我们的问题,比如我们这里想要讨论的质量相似性(多相似是相似,多大的差异我们可以接受)。
我们可以使用计算机程序来做统计,但我们也需要理解我们的用途和使用方向,如何正确地使用统计工具来获得正确的结果,是我们需要结合应用实际考虑的问题。统计工具与我们常接触的分析检测方法并无不同,粗浅地说,在执行分析检测或者统计分析时,我们都需要先理解分析检测方法和统计工具的适用对象、操作注意点,才能获得正确的结果。
统计分析工具在生物制药CMC的应用日趋广泛,不断更新的药典、指南、文献中多处提及了不同的统计分析工具在各个领域的应用,工艺表征研究(等效性检验、容忍区间)、工艺变更可比性研究(等效性检验、容忍区间)、稳定性研究(效期预测)、分析方法的适用性评估(等效性检验、容忍区间)、分析方法变更对比评估(等效性、非劣性检验)、异常值识别(Dixon法、ESD法、ROUT法等)、分析方法验证(方差成分分析、统计过程控制)等等,因此,我们应当熟悉了解这些统计工具,并结合实际工作进行使用,使其成为我们解决实际问题的利刃。
参考文献:
1.《生物类似药研发与评价技术指导原则(试行)》
2.《生物类似药相似性评价和适应症外推技术指导原则》
3.Guidance for Industry - Quality Considerations in Demonstrating Biosimilarity of a Therapeutic Protein Product to a Reference Product.
4.Guidance for Industry - Scientific Considerations in Demonstrating Biosimilarity to a Reference Product.
5.Guidance for Industry - Statistical Approaches to Evaluate Analytical Similarity (withdrawn).
6.Guidance for Industry - Development of Therapeutic Protein Biosimilars, Comparative Analytical Assessment and Other Quality-Related Considerations.
7.Meiyu Shen, Tianhua Wang & Yi Tsong (2017) Statistical considerationsregarding correlated lots in analytical biosimilar equivalence test, Journal of BiopharmaceuticalStatistics, 27:2, 213-219, DOI: 10.1080/10543406.2016.1265541.
8.Yi Tsong, Xiaoyu Dong & Meiyu Shen (2016): Development of StatisticalMethods for Analytical Similarity Assessment, Journal of Biopharmaceutical Statistics.
9.Dong X, Bian Y, Tsong Y, Wang T (2017) Exact test-based approach for equivalence test with parameter margin. J Biopharm Stat 27: 317-330.
10.Burdick RK, Thomas N, Cheng A (2017) Statistical considerations in demonstrating CMC analytical similarity for a biosimilar product. Statistics in Biopharmaceutical Research 9: 249-257.
11.Yu-Ting Weng, Tsong Y, Shen M, Wang C (2018) Improved Wald Test for Equivalence Assessment of Analytical Biosimilarity. Int J Clin Biostat Biom 4:016. doi.org/10.23937/2469-5831/1510016
12.Wang, Tianhua & Tsong, Yi & Shen, Meiyu. (2019). Sample Size Consideration for Equivalent Test of Tier-1 Quality Attributes for Analytical Biosimilarity Assessment. 10.1007/978-3-319-67386-8_3.
13.Xiaoyu (Cassie) Dong, Yu-Ting Weng & Yi Tsong (2017) Adjustmentfor unbalanced sample size for analytical biosimilar equivalence assessment, Journal ofBiopharmaceutical Statistics, 27:2, 220-232, DOI: 10.1080/10543406.2016.1265544.
14Chow SC, Song F, Bai H. Analytical Similarity Assessment in Biosimilar Studies. AAPS J. 2016 May;18(3):670-7. doi: 10.1208/s12248-016-9882-5. Epub 2016 Feb 12. PMID: 26873509; PMCID: PMC5256601.
15.FDA Application number:761099Orig1s000 Product Quality Reviews
16.https://www.fda.gov/drugs/biosimilars/biosimilar-product-information
17.Kyoung Hoon Lee, Jihun Lee, Jin Soo Bae, Yeon Jung Kim, Hyun Ah Kang, Sung Hwan Kim, So Jung Lee, Ki Jung Lim, Jung Woo Lee, Soon Kwan Jung & Shin Jae Chang (2018) Analytical similarity assessment of rituximab biosimilar CT-P10 to reference medicinal product, mAbs, 10:3, 380-396, DOI: 10.1080/19420862.2018.1433976