在2022年11月,由RDPAC 的药学团队对中美欧药学技术指导原则和指南进行了调研与对比,总结分析了中美欧指南的标准差异及实施情况差异,采用了主题词及分类方式、深度对比、报告撰写和定稿流程,最后呈现研究报告,给到药审中心参考。
2019 年 7 月 16 日,国家药监局发布了“关于进一步完善药品关联审评审批和监管工作事 宜的公告(2019 年第 56 号),该指导原则进一步完善了我国的原辅包与制剂的关联审评审批 制度。2020 年 4 月 30 日,基于新《药品管理法》和《药品注册管理办法》发布的《化学原料 药、药用辅料及药包材与药品制剂关联审评审批管理规定(征求意见稿)》,自发布以来,仍未落地实施。为进一步明确关联审评审批管理要求,解决实际工作中存在的原辅包登记及生命 周期维护等问题,建议根据业界反馈的意见和建议,尽快落实该法规,便于业界参考执行。推荐加急修订并发布,同时加强工业界培训和交流。
国外相关参考指南:
21 CFR 314.420 Drug master files - Guideline for Drug Master Files (DMF)
Type II
a) Guidance for Industry M4Q: The CTD - Quality".
b) Guidance for Industry: ANDAs: Stability Testing of Drug Substances and Products: Questions and Answers DRAFT GUIDANCE
c) GDUFA
d) ICH Guideline:Q11 Development and Manufacture of Drug Substances
Type III
a) Guidance for Industry: Container Closure Systems for Packaging Human Drugs and Biologics: Chemistry, Manufacturing, and Controls Documentation
b) MAPP 5015.5 CMC Reviews of Type III DMFs for Packaging Materials Type IV
a) DMF Guidance
b) Guidance for Industry: Nonclinical Studies for the Safety Evaluation of Pharmaceutica Excipients
Type V
a) 21 CFR 314.420(a)
Guidance for Industry for the Submission Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products
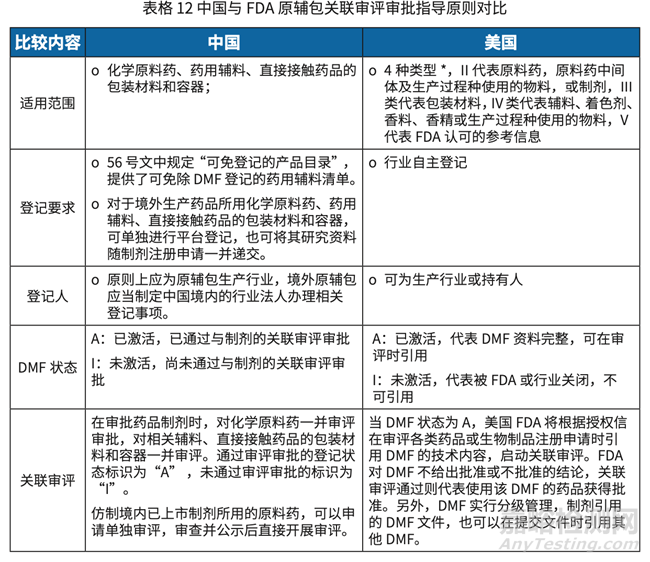

• 中国实施实践问题:
美国 DMF 适用范围较宽泛,比如 II 类除了原料药外,还包含原料药中间体以及生产过程种使用的物料或制剂。在我国,原料药中间体、制剂中间体,混粉及生物技术原 料药等,仍未纳入登记范围及关联审评审批管理。
在原辅包产品进行登记时,同一申请人 / 登记人的多个生产厂生产的同一产品或同一 产品的不同级别,目前不能公用同一个登记号。
2020 年 4 月 30 日发布的关联审评审批管理规定,原辅包行业授权使用书模板中要求 提供给药途径,且目前是一个产品使用一份行业授权使用书。
我国法规规定,原辅包登记人应为原辅包生产行业,美国登记人可为生产行业或持有人, 且允许 DMF 之间进行转让。
相较于美国非活性成分 ( 辅料 ) 数据库,我国对于原辅包登记相关信息公开内容无原 辅包的等级和用途,此信息的公布有利于制剂行业在选用原辅包时了解更多信息。
目前法规体系中化学药品和生物制品的上市后药学变更技术指导原则,主要针对已上 市的化学药品,化学原料药及生物制品,但无针对辅料和药包材相关技术指导原则, 相关变更无参考法规。
涉及原辅包来源变更的补充申请的审评时限不明确。根据 2020 年 4 月 30 日发布的关 联审评审批征求意见稿第 33 条规定“对于药品制剂变更药用辅料和药包材来源的补 充申请,药用辅料和药包材未通过技术审评的,审评时限为 130 个工作日”,而根据《药 品注册管理办法》规定,上市后补充申请的审评时限为 60 个工作日。
根据目前要求和实际,未登记或登记状态为 I 的辅料包材变更,制剂上市许可持有人均应 通过补充申请提交,此类申请可能会触发注册检验。
• 相关的国内监管法规:
1. 2015 年国务院发布 44 号文,尝试药用辅料、药包材与制剂一并审评审批。
2. 2016 年,总局分别发布了 134 号和 155 号文,取消了辅料注册批件,启动了关联审评 审批,并规定了辅料包材关联审评审批资料要求。
3. 2017 年,总局发布了 146 号公告,我国开始正式实施原料药、药用辅料和药包材登与 制剂记和关联审评政策,并由国家食品药品监督管理总局药审中心建立原料药、药用 辅料和药包材登记平台与数据库。
4. 2019 年,国家药监局发布了《关于进一步完善药品关联审评审批和监管工作有关事宜 的公告》(2019 No. 56),规定原辅包与药品制剂关联审评审批由原辅包登记人在登 记平台上登记,药品制剂注册申请人提交注册申请时与平台登记资料进行关联。56 号 公告根据辅料包材风险进行分级管理,并按需简化资料要求。
5. 2019 年 12 月1日,《药品管理法》生效,药品管理法第二十五条规定 “国务院药品 监督管理部门在审批药品时,对化学原料药一并审评审批,对相关辅料、直接接触药 品的包装材料和容器一并审评,对药品的质量标准、生产工艺、标签和说明书一并核 准” ,在法律层面确立了原辅包与药品关联审评审评管理地位。
6. 2020 年 7 月 1 日,《药品注册管理办法》生效,第二章基本制度和要求第十四条再次 提出国家药品监督管理局建立化学原料药、辅料及直接接触药品的包装材料和容器 关联审评审批制度,在部门规章层面确立了原辅包与药品关联审评审评管理制度。 其中第三章药品上市注册第三节关联审评审评制度,规定了原辅包与药品制剂关联 审评审批,由原辅包生产行业在原辅包登记平台登记,药品制剂注册申请人提交注 册申请时与平台登记资料进行关联;在药品制剂注册申请时,选用未登记的原辅包的, 相关研究资料应当随药品制剂注册申请一并申报。药品制剂注册申请与已登记原辅 包进行关联,药品制剂获得批准时,即表明其关联的原辅包通过了技术审评,登记 平台标识为“A” ;未通过技术审评或尚未与制剂注册进行关联的标识为“I” 。药审 中心向社会公示登记号、产品名称、行业名称、生产地址等基本信息,供药品制剂注 册申请人选择。
7. 2021 年4月,为贯彻落实中共中央办公厅、国务院办公厅《关于深化审评审批制度改 革鼓励药品医疗器械创新的意见》(厅字〔2017〕42 号)中关于药品与药用原辅料和 包装材料关联审批的要求,同时根据新版《药品管理法》、《药品注册管理办法》、
《总局关于调整原料药、药用辅料和药包材审评审批事项的公告》(2017 年第 146 号, 以下简称“146 号公告”)和《国家药监局关于进一步完善药品关联审评审批和监管 工作有关事宜的公告》(2019 年第 56 号,以下简称“56 号公告”)关于原料药、药 用辅料和药包材(以下简称“原辅包”)在审批药品制剂注册申请时一并审评审批的 要求,药审中心起草了化学原料药、药用辅料及药包材与药品制剂共同审评审批管理 规定(征求意见稿),目前尚未定稿。
除以上法规和指导文件之外,2020-2021 年陆续发布了很多公告、办法及指南,对化学原 料药登记要求,缴费,已上市药品中原辅包变更作出了相应规定。
建议:
建议扩展登记适用范围,对于原料药中间体、制剂中间体及生物技术原料药也纳入登记范围。
建议原料药的批准日期与平台转“A”的日期开始计算,此日期应与《化学原料药批准通知书》载明的日期相同。同时,建议尽快实现化学原料药批准通知书及其附件的“平台打印”功能,以便申请人 / 登记人进行化学原料药的周期维护。
按照《已上市化学药品药学变更研究技术指导原则(试行)》 第四章的要求,变更纳 入登记管理的辅料 / 药包材,变更后的辅料 / 药包材尚未登记或登记状态为 I 的,按照 重大变更管理。明确此类变更仅递交路径按照重大变更管理,但是相应的研究工作, 应依据制剂的风险评估结果及《已上市化学药品药学变更研究技术指导原则(试行)》 /《已上市生物制品药学变更研究技术指导原则(试行)》的要求提交变更支持性资料, 而非将技术要求按登记状态升级并按照重大变更管理。
当制剂申请同时关联已登记的原辅包进行关联审评时,建议原辅包 DMF 和制剂申请之 间是平行审评,对原辅包的发补应先于对制剂的发补或同时进行发补,这也是国际关 联审评审批程序的惯例,此举可使药品上市许可持有人同时准备对于药品各方面的提问,同时也可保证所有的疑虑都能及时解决。
为保证患者用药及药品的持续供应,建议根据《药品注册管理办法》规定,原辅 包的关联审评同制剂审批类变更的审评时限一致,即六十个工作日,补充申请合并申报事项的,其审评时限为八十个工作日。
建议扩展豁免辅料的范围,有些结构简单的无机盐或酸,合成工艺简单,作为 pH 调 节剂是可以豁免的,很多供应商并不进行平台登记,但因其具备缓冲作用,有时也用 作缓冲液 , 建议此种情形可按免登记进行管理。
建议考虑重新定义原辅包的平台登记规则:
(1)同一申请人 / 登记人的不同生产厂所生产的原料药、药用辅料和药包材,如果使 用基本相同的生产工艺和相同的主要质量标准,可以在同一个登记号下进行登记。 登记时,应注明所有相关的生产厂。
(2)同一原料药、药用辅料和药包材的不同质量等级,如粒径,药包材材质厚度等, 可以共用同一个登记号。质量标准可列入不同质量等级的可接受标准。
(3)相同原理生产工艺的同种活性成分,也可以根据其不同种质量等级和适用范围, 按不同登记号管理,而非只能登记一种最优工艺。例如行业有可能由于不同多晶型, 盐或水合物等的需求,而使用不同的生产工艺,其工艺并无优劣之分。
建议考虑更改行业授权书模板,纳入多个制剂产品,同时,不要求列明给药途径。
参考文献
1.国内外药品技术指导原则体系对比研究 (药学部分》,国家药品监督管理局药品审评中心,中国药品监督管理研究会,药品监管研究国际交流专业委员会,中国外商投资企业协会药品研制和开发工作委员会(RDPAC),2022年11月



