质量研究的目的是通过对影响药品质量的各方面因素进行研究,确定控制药品质量的项目、分析方法,并综合药学研究、药理毒理和临床研究的结果制定终产品的质量标准,主要关注以下几方面:
1.手性杂质的确定
在手性药物质量研究中的一个关键问题就是对药物光学纯度的控制。而影响药物光学纯度的手性杂质主要来源于两个方面:原料药的制备工艺中引入的手性原料、手性试剂、反应副产物及副反应产物等工艺杂质;原料药本身不稳定,构型发生翻转而形成的立体异构体。因此在研究之初, 就需根据原料药的制备工艺与各手性中心的稳定性确定需要控制的手性杂质。在这方面存在的一个主要误区即是:当分子中存在多个手性中心时, 不考虑制备工艺与各手性中心的稳定性情况,不加区分地只对并不可能存在的对映异构体杂质进行研究控制, 而对实际可能会存在的非对映异构体杂质却毫无分析与研究。
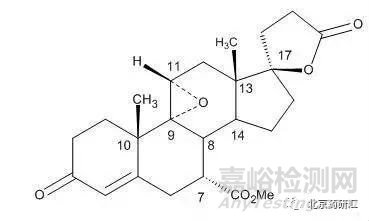
实例1:依普利酮分子中存在8个手性中心,结构如下:
由结构式可知:其中有些手性中心,如8、10、13位是处于甾体母核上,一般是天然形成的刚性结构,不太容易发生构型的翻转;而有些手性中心,如7、9、11、17则大都是后续反应中引入的,可能会产生相应的立体异构体杂质。因此,该药物中可能存在的立体异构体杂质只可能是非对映异构体,而不可能存在对映异构体杂质。
2.方法的选择及验证
目前常用于控制手性药物光学纯度的方法有两种:比旋度法与手性色谱法。影响比旋度数值的因素较多, 该数值的变化并不一定能灵敏、准确地反映出立体异构体含量的变化, 当比旋度数值较小时更是如此。例如,当采用1 dm的旋光管, 浓度为1g·100mL-1的溶液进行测定时,假设某手性药物仅含有一个手性中心, 该药物的比旋度为+100o。则根据计算公式可知:当该药物中混有1.0%的左旋异构体杂质(假设其他杂质忽略不计)时,此时药物的旋光度将由+1.00o变为+0.98o而如果该手性药物的比旋度仅为+10o, 则当该药物中同样混有1.0%的左旋异构体杂质时,此时药物的旋光度将由+0.10o变为+0.098o, 旋光度的变化值仅为0.002o。此时如仍采用药典附录中提到的读数至0.01o的旋光计, 则两者旋光度的差值超出了仪器的测量精度, 在该旋光计上将显示相同的读数, 根本反映不出产品中混有1.0%的左旋异构体杂质。这也是为什么在制定质量标准时,当手性药物在不同溶剂中的比旋度相差较大时, 一般选择具有较大比旋度数值的溶剂作为标准中测定用溶剂, 以尽可能灵敏地反映手性药物光学纯度的变化情况。
实例2:甲基多巴在0.1N HCl中的比旋度为[α]25D-5.2(C=2.0), 在水中的比旋度为[ α] 20 D-14(C=1.09), 而在4.4% AlCl3水溶液中的比旋度为-25~-28。为保证该项目更能灵敏地反映甲基多巴光学纯度的变化,在中国药典收载的甲基多巴标准中,就采用了配置较复杂的4.4%AlCl3水溶液作为测定比旋度的溶剂, 而未采用常见的0.1N HCl或水作溶剂。
另外,当手性药物含有多个手性中心时, 随着立体异构体数目的增多, 比旋度数值的变化将会受到更大的影响, 从而更加难以从比旋度的变化来获知手性药物光学纯度的具体变化情况。
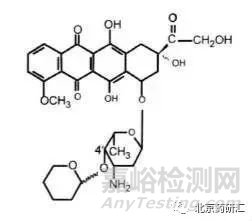
实例3:吡柔比星在4'位产生的2个差向异构体在氯仿中的比旋度是相同的,均为+150(C=0.2)。此时如果仅用测定吡柔比星在氯仿中的比旋度来控制4'位差向异构体的含量就变得毫无意义了。因为不论该差向异构体的含量如何变化,所测定的比旋度均不变。
基于以上原因,手性色谱法可以更为准确直观地反映各立体异构体杂质的变化情况。但是,在质量研究中同样需要对所采用的手性色谱法进行充分的方法学验证, 以确保该方法确实能够以足够的灵敏度检测到需控制的立体异构体杂质。为此, 在验证时需在以下两方面加以注意:分析确定需控制的立体异构体杂质;尽量采用杂质对照品进行专属性和定量限的验证。在分析确定需控制的立体异构体杂质时, 首先应根据原料药的制备工艺, 分析工艺中可能产生哪些立体异构体杂质;其次, 需要设计一系列的降解试验来考察确定分子中各手性中心构型的稳定性, 以分析确定该手性药物在哪些因素作用下可能降解产生哪些立体异构体杂质。这样,在分析方法验证时, 就能目的明确地考察该分析方法是否能分离并检测出所有可能存在的立体异构体杂质。
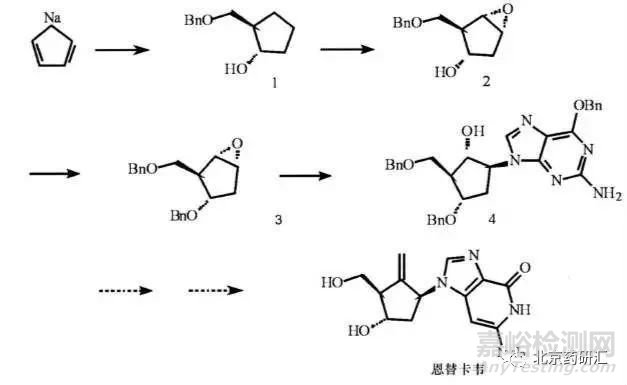
实例4:恩替卡韦
抗乙肝病毒药物恩替卡韦的合成,在引入手性中心的第1, 2, 4 步反应中, 只要这些反应不是采用立体专属性的反应, 以百分之百地得到所需构型的反应产物, 且后续操作中不能有效地去除所产生的立体异构体杂质, 则终产品中就可能混有恩替卡韦的所有其他7个立体异构体杂质。除非在反应过程中已采用合适的纯化手段去掉各步反应中引入的立体异构体杂质,并采用专属有效的分析方法对各步反应的中间体中的立体异构体杂质进行监控, 否则在终产品的质量控制中,就应采用合适的分析方法对这7个立体异构体杂质进行控制。此时终产品的质控难度是非常大的。
由此例可见,了解手性药物的制备工艺, 并通过对引入手性中心的化学反应的立体选择性进行分析, 可以全面了解工艺中可能引入的立体异构体杂质,从而采取合适的分析手段分别在工艺过程中或终产品中进行控制质量标准的制定,在制定手性药物的质量标准时一定要结合手性药物的特点,重点关注能否切实控制手性药物的光学纯度。对于合成制备的手性原料药,由于合成过程中会引入各种手性杂质,并且在放置过程中也可能会因为构型不稳定而降解产生相应的立体异构体杂质,所以原料药的质量标准中一般均须设置专属而灵敏的立体异构体杂质检测项目。
对于发酵或提取制备的手性原料药, 由于各手性中心均是天然形成的,一般都是立体专属性的,产生立体异构体杂质的可能性不大,如果有充足的文献或实验依据证明各手性中心在放置过程中构型是稳定的,则可以仅用比旋度项目来粗略控制该手性药物的光学特性。
对于制剂而言:如果手性原料药在放置过程中构型是稳定的, 且原料药中所含立体异构体杂质不具生理活性,则制剂的质量标准中可不纳入立体异构体杂质控制项目。但是,随着研究的深入,越来越多的已上市消旋体药物被开发成单一的立体异构体药物。
实例5:在苯磺酸氨氯地平片的基础上进一步开发了苯磺酸左旋氨氯地平片。两者的区别仅在于后者的规格较前者小一半。此时如果不根据苯磺酸左旋氨氯地平片的研究结果,在质量标准中酌情制定立体异构体杂质检查项或立体专属性的鉴别项,则在质量标准中很难区分消旋体与左旋体片剂。因此有必要在单一的立体异构体制剂的质量标准中至少订入立体专属性的鉴别项,以利于上市药品的监督管理。
3.稳定性研究
手性药物在一定条件下,手性中心可能会发生构型反转,从而生产相应的立体异构体。正是由于构型有可能发生翻转,所以在手性药物的稳定性研究中就应该采取有效的分析方法监控各手性中心构型的稳定性。在此需注意以下几方面的问题:
① 如前所述, 基于比旋度法的局限性, 最好采用更为灵敏的手性色谱法来监测各个立体异构体的变化情况。
② 要注意考察制剂中的手性药物构型是否仍然稳定, 尤其是一些液体制剂。
③ 要根据手性中心的多寡与各自的稳定性, 选择合适的立体异构体进行检测。
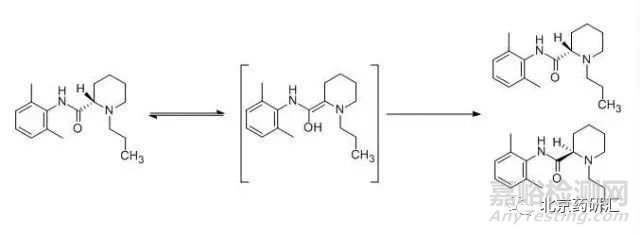
实例6:在长效酰胺类局麻药--甲磺酸罗哌卡因的质量标准中,为检测手性HPLC法的系统适应性,取甲磺酸罗哌卡因对照品适量,加水制成每1mL含75μg的溶液,于90℃放置6h后,即可得到含有0.5%至1.0% R-(+)对映异构体的对照溶液。由此可见,甲磺酸罗哌卡因分子中的手性中心在溶液中加热即可发生构型反转而生成其对映异构体。
由罗哌卡因的结构可知,手性中心的邻位有吸电子的羰基,从而使手性中心上的氢成为活性氢,部分活性氢原子在溶液中获取一定的能量即可以氢离子的形式脱去,使手性中心成为平面结构的负碳离子,根据进攻方向立体位阻的大小,溶液中的氢离子再以一定的比例从平面的上、下两个方向进攻该负碳离子,从而分别得到甲磺酸罗哌卡因及少量的右旋体杂质。
实例7:二氢胆固醇构型的转换
二氢胆固醇在碱的催化下, 其3位的α构型的羟基会部分转化为β 构型,从而产生非对映异构体。反应示意图见图5。故在此情况下, 稳定性考察时监测的重点降解产物就应该是非对映异构体, 而不是对映异构体。
此外,在考察手性药物构型的稳定性时, 考察条件不能局限于国内稳定性研究指导原则通常所采用的影响因素试验的条件。例如, 前面提到的甲磺酸罗哌卡因, 如采用一般的影响因素试验条件, 就可能观察不到构型的翻转现象。
综上所述, 在手性药物的药学研究中, 一定要结合手性药物的特点开展相应的研究,否则所作的研究难以真正反映手性药物的稳定性,也难以准确控制其光学纯度。另外,在具体的研究过程中,也不能拘泥于指导原则中的方法机械地执行。而是要在遵循其基本原则的基础上,根据具体情况灵活运用。应注意根据手性药物的处方与制备拘泥工艺、手性中心的多寡与构型的稳定性、各立体异构体杂质生成的可能性与毒性等情况,采用合适的分析方法分别从起始原料、工艺过程与终产品的质量标准等各个方面对手性药物的光学纯度进行全程的监控,以充分保证手性药物的质量是稳定可控的。