创新医疗器械的设计输出往往和传统产品按部就班的进行有很大的差别,通常我们需要进行多次设计更改和动物试验才能够将产品最终设计定型。因此创新医疗器械的设计输出就比较复杂了,在此今天给大家介绍下创新医疗器械的设计输出--前期个人经验供大家参考!
1.主要任务 Main Task
1.1产品设计及输出 Product Design and Output
本阶段应结合设计输入的要求,对产品进行整体设计。针对结合在一起形成产品的所有子系统、零件、组件和原材料的设计输出应形成文件,这些文件包括:产品图纸(包括原材料、零件、组件、半成品或成品图纸),适当时,建立原材料质量标准(适当时输出原材料规格说明书)。适当时,应输出设计产品设计方案且应经过评审,评审结果应形成文件。
1.2过程设计及输出 Process Design and Output
1.2.1工艺研究与输出Process Research and Output
本阶段应结合设计输入的要求,对产品工艺进行设计,适当时输出工艺开发方案且应经过评审,评审结果应形成文件。适当时,应分析和确定本阶段需完成的工艺研究实验,输出先行试验报告。对于无菌提供产品,至少应完成内包装、灭菌等工艺与现有工艺等同分析或工艺研究报告或过程确认。如无等同性,则应在设计输出评审前完成。本阶段的过程确认包含IQ和OQ,且应在设计输出评审前完成。
1.2.2过程设计与输出Process Design and Output
过程设计输出是针对产品制造、维修、服务、环境、库存、运输等制造程序的输出,本阶段应输出产品制造过程文件,包括:工艺流程图、工艺规范、生产批记录表、设备规范等。适用时,工艺规范包括:生产工装图纸、标准操作规程、工艺参数表、电子程序等。适用时,设备规范包括:生产设备及清单、生产工夹模具及清单、设备操作规程及维护保养规程。
1.3生产辅料设计及输出 Design and Output of Production Auxiliary Materials
本阶段应对重要度为A、B类的生产辅料进行设计,并输出可接收的质量标准。
1.4标准测试方法Standard Test Method
需要建立标准测试方法(STM)时,应优先分析和采用法规/标准(如:ASTM、ISO、GB、YY等)中的方法。非法规/标准中的测试方法应在设计验证完成前(注册送检前)完成方法学验证。适用时,本阶段应输出相应的检测工装图纸、检测设备及工装夹具清单。
1.5设计评价 Design Evaluation
产品设计评价应识别本阶段需要采用的产品设计评价方式,可以包括:原样研究、早期性能及指标研究、可行性动物试验研究、早期生物学评价研究、早期可行性临床研究、设计定型验证、工艺研究、以及可用性的形成性评价等方面。依据产品规范中所列各项性能,就上述各评价方式的选择进行判断,输出产品设计评价方案和报告。
1.5.1原样研究 Research of original Samples
原样研究为验证关键性能是否满足要求。原样应经过评审,评审要求按照技术评审方式进行,通过该评审是产品设计/过程设计进一步开展的前提条件。
本阶段应对原样进行相应的测试,测试至少包括关键物理性能指标、功能实现、可用性评价等,输出原样测试报告。适用时,本阶段应输出先行试验报告,其至少应包括试验目的和意义、适用范围、性能指标测试结果、结果分析、稳定性分析等。样品或样机生产及检验的可追溯性。
当产品的材料包含可降解材料或潜在降解材料时,应在本阶段应完成产品储存环境研究和早期产品加速老化研究,并输出研究报告。
1.5.2早期性能及指标研究与输出Early performance and index research and output
本阶段活动包括测试方法研究、产品与对比产品性能对比评价和产品性能及指标制定依据研究。
当产品为无菌医疗器械或微限医疗器械时,注册检验送检前应完成微生物、无菌方法学确认;当为EO灭菌时,应在注册送检前完成EO残留方法学确认。
当产品为免临床目录内产品或通过同品种比对豁免临床试验时,应进行产品与对比产品性能对比评价。该评价应至少包括:目的、检验设备要求、样品信息、对比项目及检验方法、测试结果、结果分析等。该评价应在设计输出评审完成前完成。产品与对比产品性能对比评价结果是产品性能及指标制定依据的输入来源。
应识别本阶段产品性能及指标制定依据研究方法,可以包括引用标准、文献分析、产品与对比产品性能测试对比、产品实际测定结果等。在注册送检前应输出产品性能及指标制定依据研究资料,同时更新设计输入。
1.5.3可行性动物试验研究与输出Feasibility animal experiment research and output
可行性动物试验样品可使用未定型产品。适用时,本阶段应基于非临床研究文献,输出可行性动物试验研究方案,应至少包括实验目的、受试器械、试验用动物、动物模型选择依据、动物数量、观察时间、评价指标、偏差信息等。试验结束时,本阶段应形成可行性动物试验研究报告,应至少包括信息参照2021年国家药监局关于发布医疗器械动物试验研究注册审查指导原则《医疗器械动物试验研究注册审查指导原则第二部分:试验设计、实施质量保证》。
适用时,非临床研究文献应进行汇总形成文件(用于注册资料储备)。
1.5.4早期可行性临床研究与输出Early feasibility clinical research and output
关于早期可行性临床可行性在前期撰写过一篇医疗器械研发能否开展小样本临床试验,详细的可以点击蓝色字体进行了解。
适用时,本阶段应基于临床研究文献,输出可行性临床研究方案,应至少包括方案摘要、申报者信息、临床研究机构和研究者列表、临床试验目的和项目内容、临床研究的背景资料、产品的特点结构组成、产品的适应症与禁忌症、总体设计、数据管理等。
适用时,临床研究文献应进行汇总形成文件(用于注册资料储备)。适用时,早期可行性临床研究应进行且输出至少3-5例符合预期用途要求的表明产品有效性的临床数据结果。
1.6包装/标签设计与输出 Packaging/Label Design and Output
应在本阶段输出包装设计、工艺和接收的质量标准(如图纸)。
1.7进货/过程/半成品/成品检验规程 Receiving/ In-Process/ Semi-finished/Product
针对产品制造过程的检验要求应形成文件,输出进货、过程、半成品或成品检验规程及其空白检验记录单。
1.8 设计定型及输出 Design Freeze and output
设计定型验证用以支持设计定型,设计定型验证评价包括:产品性能设计定型、产品工艺设计定型。本阶段应输出支持设计定型验证的DMR清单,包括文件编号、文件版本号。适用时,产品性能设计定型应进行早期可行性临床研究(详见1.5.4)以确认产品性能设计定型;适用时,产品工艺设计定型应进行设计定型样品的生产,并按照技术要求的项目进行全性能检测。在进行产品工艺设计定型样品生产前,应输出物料清单(BOM)。
适用时,本阶段应输出注册送检典型型号规格研究资料,用于工艺设计定型样品的规格型号。本活动应经过评审,通过是进行工艺设计定型验证的前提条件。工艺设计定型样品的样本规格和所用DMR文件应与注册检验的原则保持一致,且数量应不小于10套。
设计定型验证应经技术评审即设计定型评审,并输出设计定型报告,包括本次验证所对应样品的设计输出及生产过程文件清单及版本。设计定型评审是开展设计输出评审的前提条件。
1.9产品说明书/技术要求Instructions for Use/Technical Requirements
完善产品技术要求,产品说明书等技术文件。完整的产品使用说明书分为产品使用说明书内容和包含产品使用说明书排版在内的最终整合两项活动。排版的产品使用说明书,需临床试验的产品在设计验证完成前输出,其他产品在设计确认完成前输出。最终销售使用的产品使用说明书版本应在获得注册证后输出。同时注意在各个阶段更新该文件。
应识别本阶段产品性能及指标制定依据研究方法,并在设计定型后输出产品技术要求。研究方法包括,引用标准、文献分析、产品与对比产品性能测试对比、产品实际测定技术水平等。结果应形成文件,同时更新至设计输入。必要时,进行产品与对比产品性能对比评价。
1.10材料评价Material Evaluation
本阶段应根据“物料清单和原材料质量标准”等信息,制定“材料评价计划”,材料评价包括原材料的安全性评价资料、物料小样确认评价、小批量材料评价(包括多批次评价)、材料的生产适用性评价(来源归属类别为物料类的偏差)以及材料对产品性能影响的评价。适用时,原材料的安全性评价资料包括:MSDS、生物学试验报告、化学成分检测报告、供应商生产资质、采购合同、质量协议、进货检验表等。
根据材料评价计划,设计定型评审前输出原材料的安全性评价资料、物料小样确认评价。
小批量材料评价(包括多批次评价)、材料的生产适用性评价(来源归属类别为物料类的偏差)以及材料对产品性能影响的评价应在应在设计验证完成(T5)并形成材料评价报告。
1.11风险管理Risk management
1.11.1设计故障模式与效应分析(FMEA)
利用故障模式与效应分析(FMEA)方法分析产品设计的风险,本阶段应识别故障模式、故障原因、故障结果、探测方法,进行风险估计和风险评价,适用时制订风险控制计划,输出设计故障模式与效应分析报告(DFMEA)。
1.11.2过程故障模式与效应分析(PFMEA)
利用故障模式与效应分析(FMEA)方法分析产品制造过程的风险,本阶段应识别故障模式、故障原因、故障结果、探测方法,进行风险估计和风险评价,适用时制订风险控制计划,输出过程故障模式与效应分析报告(PFMEA)。
1.12(必要时)专利申请 (If necessary)
根据产品设计或工艺设计情况,必要时进行专利申请。
1.13生物学评价 Biological evaluation
本活动适用于与人体直接接触和/或间接接触的产品。
1.13.1生物学评价计划 Biological evaluation Plan
本阶段应输出生物学评价计划,设计验证完成前完成生物学评价报告。生物学评价应按照ISO 10993-1:2018的要求确认生物学测试项目。生物学评价计划应形成文件,至少包括:生物学终点测试项目、各项目的明确途径、试验方法、样本量需求数量、试验周期、获得报告时间等。
生物学终点测试项目应根据“注册路径”,按照ISO10993-1:2018或GB/T 16886.1-2022的要求结合医疗器械分类及接触类型选择。
1.13.2生物学试验样品 Biological test samples
原则上生物学试验样品应采用终产品。适用时,应输出生物学试验样品生产方案。适用时,应明确采用半成品的原因和依据。适用时,应输出典型型号规格和样品制备说明。
最后筛选符合资质的检测中心,按照检测中心要求将送检资料和生物学试验样品一并送至检测中心。在设计验证完成前应获得生物学试验报告。
1.14 设计验证策划 Design Verification Plan
本阶段可根据设计输入要求”输出“设计验证追溯表”,设计验证项目应追溯到“设计输入”的全部要求,设计验证完成前更新追溯表,并增加设计验证结果。
1.15设计转移策划 Design Transfer Plan
本阶段应输出设计转移计划,该计划应至少包括以下内容:
转移小组;
接受标准;
统计学方法、样本量信息;
典型性样品选择依据;
过程能力验证方式(试生产或评估);
转移小组各职能人员职责。
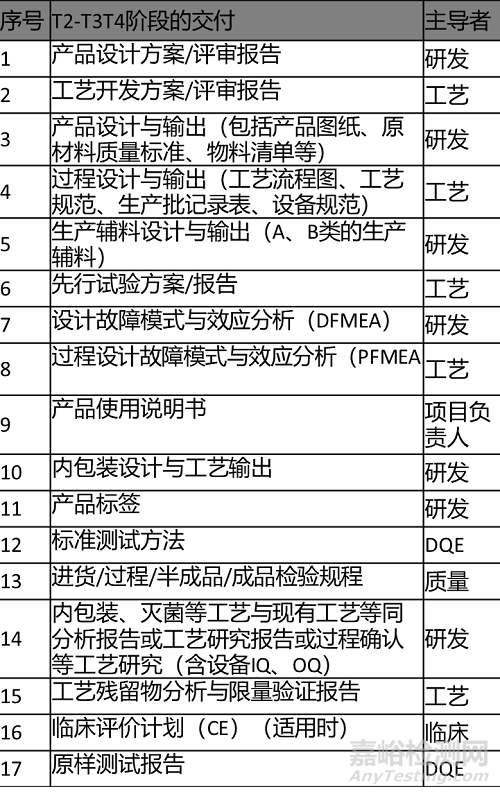
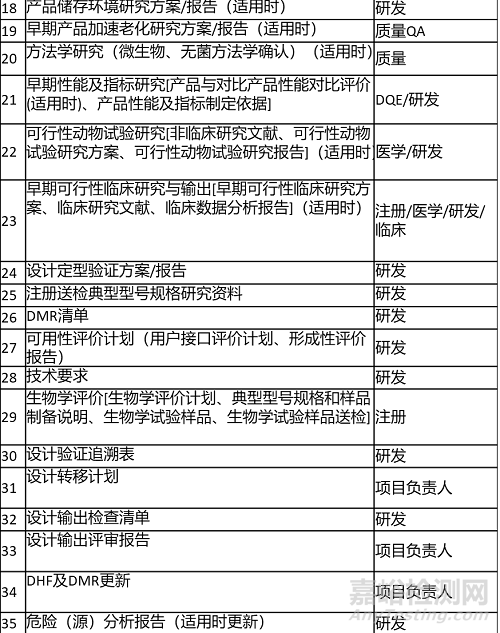
2.输出清单 Output List
输出内容及清单如下表所示。
3.设计输出评审 Design outputs review
根据策划阶段形成的检查清单对输出的适宜性和完整性进行评审。