摘要
目的:分析«美国药典»药包材标准体系概况及其最新进展, 为我国药包材标准体系的建立与优化提供参考。
方法:对«美国药典»药包材标准体系框架结构与标准内容进行解析, 对药包材产品标准和方法标准、美国药包材管理法规的相互关系进行比较分析。
结果:«美国药典»药包材标准体系相对完善, 药包材评价标准具有鲜明特色, 药包材标准贯穿药包材使用全生命周期, 同时药典论坛定期更新。
结论:«美国药典»相对完善的药包材材质标准, 产品标准与评价标准相互支撑并贯穿药包材使用全生命周期, 都对我国药包材标准体系建设具有重要参考价值。
关键词
中华人民共和国药典; 美国药典; 药包材; 标准体系
正文
药包材的质量、安全、使用性能以及药包材与药物之间相容性对药品质量有着十分重要的影响。«中华人民共和国药品管理法»[1]指出, 药包材应当符合药用要求, 符合保障人体健康、安全的标准。«国家药监局关于进一步完善药品关联审评审批和监管工作有关事宜的公告»(2019年第56号)[2]要求药包材登记资料中需提供该药包材及各组件被国家标准及国内外药典、以及相关国际标准收载的信息。国内外药典药包材标准是我国药品与药包材关联审评重要依据之一。
目前, 影响较大的国外药典如«美国药典»(United States Pharmacopeia, USP)、«欧洲药典»(European Pharmacopeia, EP)、«日本药局方» (Japanese Pharmacopeia, JP)等均已收载药包材标准。
美国食品药品管理局(FDA)对药包材采取药品主文件(drug master file, DMF)的管理模式。DMF是指向FDA提交的一组文件, 共分为Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ类5种(Ⅰ类DMF于2000年停止)。其中, 包装材料为Ⅲ类DMF。FDA的DMF是关联审评的备案管理模式, 我国药包材也采用关联审评的备案管理模式, 因此研究USP药包材标准体系架构对研究制定中国药包材标准具有重要借鉴意义。笔者在本文针对USP[3]的包装标准体系及其进展进行概述和分析, 以期使我国药品和药包材研发、生产、使用、检验、审评和监管各方更好地了解USP药包材标准概况, 为关联审评和国家标准体系构建提供参考和借鉴, 以期使«中华人民共和国药典»药包材标准能够更好地服务于审评审批和上市后监管, 能够更好地推动药包材及药品行业高质量发展[4]。
1、USP组织架构及药包材标准制修订
1.1 USP组织架构
第一版USP发布于1820年。美国药典大会为USP的管理机构之一, 通过每5年一次的大会, 大会成员确认USP决议, 并选举USP理事会和领导USP标准制定专业委员会的专家委员会。理事会对组织起着至关重要的领导作用。理事会受托管理USP, 负责信托监督、全面声誉风险管理以及与其他USP管理机构合作的治理活动; 专家委员会是监督USP科学和标准制定决策的机构。成员由USP大会成员在每5年一次的会议上选举产生。专家委员会是监督USP科学和标准制定决策的机构。2020—2025年, USP共设立了专家委员会29个, 由来自41个国家和地区的996位专家组成(2021Annual Report)。其中药包材标准由通则-包装与流通(General Chapter-Packaging and Distribution, GCPD)专家委员会负责。
1.2 USP制修订流程
USP标准的制修订可由USP员工、监管方、企业或学术机构提出, 在提出的同时向USP递交支持性文件(修订原因、技术资料、方法学数据等), USP相应科学联络官对资料审核后提出标准草案, 草案经审核在药典论坛(pharmacopeial forum, PF)上发布, 向公众征求意见, 根据公众意见对标准做相应、适当的调整, 最后标准经相关专家委员会以投票方式(以多数票通过后)生效。
1.3 USP版本更新
USP自2021年起不再保留整套药典的版本号, 而是引入了以文件为中心的方式。无论单个文件是否正式生效, 都不会再被关联至一个具体的药典版本, 而是基于单独的标准本身, 从而使标准的制修订能更加及时、快捷[5]。USP是全球第一个采用单个文件为中心的药典标准, 标准更新将不再受限于药典发布周期, 而是依据标准制修订本身。
2、药包材标准体系主要内容
2.1 USP药包材标准框架
USP的药包材标准框架体系, 包括以材料及其容器为主线的通用性标准, 如玻璃、塑料、橡胶材料和(或)其成品的通用要求, 同时涵盖了满足通用要求评价的性能测试方法。除此之外, USP还设立了更为广泛的评价和研究指南性通则[6]。
USP凡例介绍了美国药典/国家处方集(United States Pharmacopoeia/ National Formulary, USP/NF)的解释和应用的基本假设、定义和默认条件。另外, 除各论中另有规定外, USP涉及所有产品都要符合<659>中规定的包装和储存要求。
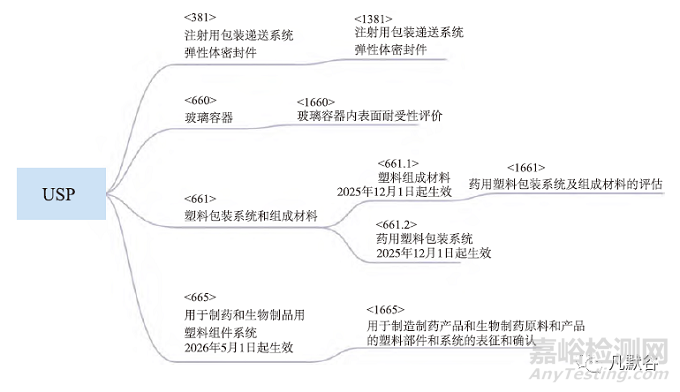
USP药包材标准体系中涉及的相关标准, 按照其分别起到的作用, 大致可以分为4类(表1)。第1类是产品通则, 包括<381><660><661><670>等。第2类是药包材通用要求, 如<659>等。第3类是性能测定标准, 包括保护性功能性生物反应性性能等的测试方法, 如<87><88><382><671>等。第4类属于通用信息, 对涉及不同种类药包材的使用评价完整性评价功能性评价相容性研究评价规范等内容加以说明, 包括<1031><1381><1382><1178><1207><1660><1661><1663><1664>等。此外, 值得注意的是, 第4类标准是非强制性标准。
2.2 USP药包材标准内容
USP中涉及药包材的相关章节分布于通用章节(general chapters)中, 其中又分为通用检查与测定法(general tests & assays)和通用信息(general information)章节。通用检查与测定法章节一般属于强制执行的内容(除非另有规定), 包括了对药包材的通用要求, 以及塑料材料及包装系统、弹性体密封件、玻璃容器和辅助组件的产品标准, 还包括对药包材功能性的测定方法。通用信息章节主要提供对于药包材生产、使用和评价方面的一些指导性意见, 属于信息类非强制性标准。
对于弹性体密封件, <381>标准提供了注射用弹性体密封件材料的方法及规范。内容包含引言、范围、试样、测试步骤(包含生物反应性、物理化学实验、功能性的程序和要求)。USP<1381>主要阐述USP<381>的应用及适用性, 在USP<381>中提供的基线要求之外, 弹性体需要符合药品质量风险水平的预期用途及弹性体成分的评估。此外, 值得注意的是将于2025年12月1日生效的USP<382>已公开发布, 在<382>正式生效之前, 使用<381>中描述的弹性组件功能适用性测试范围。而注射产品用弹性体密封件包装/输送系统功能适用性的检测方法及评估方法可以参考<1382>。
对于玻璃容器, <660>对安瓿、注射剂瓶、注射器等玻璃容器提供了检测方法及规范。<660>内容分为定义、特定测试(包含颗粒耐水性、内表面耐水性, 表面腐蚀测试)、杂质(砷), 功能性测试(有色玻璃透光率)等。此外<1660>提供了玻璃容器的类型、生产过程、内表面耐受性影响因素, 同时推荐了内表面耐水性的评价方法。

对于塑料包装系统, <661>为用于药品包装(药品、生物制剂和膳食补充剂)的塑料制品(材料、组件和系统)提供检测方法及规范。<661>内容包含引言、聚乙烯容器、聚丙烯容器、聚对苯二甲酸乙二醇酯容器、测试方法(多重内表面反射、热分析、生物测试、理化测试)等内容。未来, USP塑料包装系统将目前的<661>章节细化分为两个子章节分为章节<661.1><661.2>, 对塑料的组成材料、塑料包装系统分别建立质量标准, 新的章节将在2025年12月1日正式生效, 同时<661.1>除原有的聚乙烯、聚丙烯、聚对苯二甲酸乙二醇酯外, 涵盖了更多的材料类型, 包含环烯烃聚合物、聚酰胺6、聚碳酸酯、聚乙烯、乙烯-乙酸乙烯酯共聚物、聚丙烯、聚氯乙烯、塑化聚氯乙烯等。<1661>为通用信息类章节, 是配套<661>建立塑料材料和容器的评估方法, 同时为<661.1><661.2>的提供补充信息和应用说明。
<665>为生产用塑料组件和系统提供评价方法, 将于2026年5月1日生效。USP<1665>介绍了塑料组件系统在制药和生物制品中的评估应用方法。但值得注意的是, <665>虽为USP<1000>以下章节, 但为非强制性标准。
对于生物相容性, <87>主要描述了体外生物反应性实验的检测方法, 以用于确定直接或间接接触弹性体、塑料和其他高分子材料或从被测材料制备的特定提取物接触后的生物反应性。<88>主要描述了体内生物反应性试验的检验方法(包含全身注射试验、皮内试验、植入试验等)。<1031>主要以USP<87><88>的试验指导为基础方法, 为评估药物容器、弹性密封件、医疗器械和植入物生物相容性研究评价提供指导。
USP中其他方面, <659>提供了包装定义、辅助包装信息和与有效成分、辅料和医疗产品(如药品、组合产品, 以及标签和为符合USP要求的膳食补充剂)贮运相关的包装、辅助包装信息和贮存条件的定义和要求。<670>为用来支持或增强容器封闭系统的辅助包装组件, 这些组件包括但不限于制药线圈和容器用干燥剂等。<671>主要提供用于固体口服制剂和液体口服制剂的包装系统功能特性的标准。<1671>配套<671>主要提供了塑料包装固体口服制剂水蒸气透过率的评价方法, 包含干燥剂法和水作为替代灌装溶剂的方法, 同时提出了再包装的相关评价指南及术语定义。
<1178>提供指导口服固体药品的重新包装, 包含从原始容器封闭系统中取出药物, 并将它们重新包装到不同的容器封闭系统中进行销售和(或)进行分发的相关要求和定义。
<1207><1207.1><1207.2><1207.3>阐述了无菌产品包装完整性评价的指导原则, 包括泄露原理、无菌环境的控制、泄露测试技术、密封质量测试技术等, 同时也对相关的专业术语进行定义。
<1663>阐述了药用包装/给药系统相关可提取物方法的建立及评估, <1664>阐述了药用包装/给药系统相关浸出物的评估方法。此外<1664>的子章节<1664.1>主要阐述经口鼻吸入给药制剂中可浸出物的评估方法以及一些特定术语的规定。
2.3 USP药包材标准内容分析
FDA药包材指南—容器密封包装系统指南[3]是FDA药包材工业指南, 对药包材药学研究资料提出了框架指引, 对不同类型的包装系统基于药包材包装制剂的风险程度和药包材与药品相互作用的可能性, 提出了“四性”(即保护性、功能性、安全性和相容性)评价等相关要求, 为备案及审评提供了重要参考。USP通过强制性和信息性的药包材标准章节的收载对FDA药包材指南—容器密封包装系统指南[7]对药包材的“四性”从药典标准层面有着较好的标准对接。
关注材料容器系统的化学安全性(图1)和生物安全性: <660><661>关注材料本身的类型, 塑料制品通过红外光谱(infrared spectrophotometry, IR)和差示扫描量热法(differential scanning calorimetry, DSC)进行鉴定和表征, 玻璃通过颗粒法耐水性进行鉴别分类。<381><660><661>(塑料、弹性体密封件、玻璃材料及容器)关注物理化学测试, 结合产品自身的特点和包装药品剂型的风险程度, 设计关键质量属性的项目。<1381><1660><1661>提供塑料、弹性体密封件、玻璃化学评估的方法与要求。USP中<1381><1660><1661>3个章节对材质的化学安全性评价提供了评价方法标准。同时, USP关注材料容器系统的生物安全性: <1031>提供材料浸出物生物相容性评价指南, <87><88>等提供配套检测方法。
图1 USP 材料与材料评估药包材标准框架
Fig.1 Framework of USP pharmaceutical packaging material standards about material and material evaluation
关注材料容器系统的功能性评价: 对于弹性体密封件在目前现行有效的USP<381>中涵盖穿刺力、穿刺落屑、自密封性3个质量控制项目, 但将于2025年12月1日生效的USP<382>将对于USP<381>中涉及的功能性评价内容单独成章, 标准项目涵盖包装/交付系统完整性、针头、针尖功能适用性、柱塞功能适用性、柱塞功能适用性、护帽功能适用性等。同时, <1382>阐述了弹性体密封件功能适用性评估方法。
关注材料容器系统的保护性: 对于无菌产品的完整性, <1207><1207.1><1207.2><1207.3>系列标准为无菌产品完整性评价提供检测方法选择和验证、检测技术等指引。而塑料容器及其部件的功能性的标准和检验请参见容器性能检验<671>, 对于塑料材质功能适用性USP<671>主要提供了用于固体、液体口服剂型的塑料包装系统的水蒸气透过率和包装系统透光率测试等的标准及方法。
关注材料容器系统的相容性: <1663><1664><1664.1>对药品包装系统的可提取物、浸出物、经口鼻吸入给药制剂的浸出物提供研究评价指南。
3、USP最新进展
USP论坛(pharmacopeia forum, PF)是USP标准建立与修订的在线信息发布平台。新标准或修订标准在正式生效前, 通常以提案形式在PF上公示, 每两个月就拟增修订药典标准公开征求意见, 并一般开放90d评议期接受公众评议。近2年来药包材标准内容陆续更新(表2)。PF有效促进了标准的沟通交流与社会共同参与标准制修定。其中<383>为首次修订, 为用于药品包装和生产部件的硫化硅橡胶弹性体提供技术标准, 包含鉴别、理化检查、生物反应性等项目。
4、讨论
我国正在加快推进«中华人民共和国药典»药包材标准体系的构建, 在药包材标准的制定中, 充分发挥药典标准法典的核心作用, 补齐药典药包材标准的短板[4], 以下这些方面都可以学习借鉴USP的药包材标准内容。
我国正在加快推进«中华人民共和国药典»药包材标准体系的构建, 在药包材标准的制定中, 充分发挥药典标准法典的核心作用, 补齐药典药包材标准的短板, 以下这些方面都可以学习借鉴USP的药包材标准内容。
同时, 我国已加入人用药品技术要求国际协调理事会(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, ICH), 已实施和正在制修订的ICH相关指导原则均将逐步转化落地。因此, 未来«中华人民共和国药典»相关章节与ICH的协调势在必行, 相关研究和积累也将迫切重要。其中, ICH Q3是一个非常重要的章节, 主题是关于杂质研究, 目前ICH有4个章节分别为ICH Q3A、Q3B、Q3C和Q3D。对于包装材料杂质相关领域, ICH Q3E药品和生物制品的可提取物浸出物控制与评估于2019年6月由ICH大会批准, 并正在逐步建立过程中, 杂质检测指南计划2025年5月完成Step4通过最终指导原则[8]。ICH Q3E章节的建立将规范可提取物浸出物控制与评估方法。而对于可提取物浸出物“现有指南、药典和其他涉及可提取物和可浸出物的标准之间缺乏一致性、共识和明确性[9]。”对比各国药典, 目前只有USP在<1663><1664><1664.1>等章节详述了可提取物浸出物评估方法。可借鉴USP, 加强研究可提取物浸出物评价方法标准积累与构建, 对接ICH Q3E[10]出台, 未来逐步完成药典方法的转化。
最后, 目前USP药包材标准主要集中在玻璃、塑料、橡胶3类材质, 金属药包材涵盖高、中、低风险药品剂型, 包含保障药品保护性的辅助包装组件等。金属类药包材标准研究基础弱, 体系不完善, 但也是近期各国药典关注的重点领域之一。2021年, 中美药典基于互信、平等、务实的合作理念建立了金属类药包材标准工作组, 双方专家共同探讨并推进金属类药包材标准的建立[11], 通过公开征集参与单位[12]、征求意见建议[13]等方式, 已初步确定«中华人民共和国药典»金属类药包材标准体系规划。未来, 借鉴USP逐步完善药包材标准体系的建设, 夯实不同材料的通用要求, 尝试建立«中华人民共和国药典»金属类药包材通则, 药典药包材标准从跟跑到并跑, 并尝试逐步领跑, 建立起由塑料、橡胶、玻璃、金属的通用要求, 延伸到对陶瓷、纸类等药包材的标准, 借鉴国内外经验, 立足我国国情, 进一步完善药包材标准框架体系, 构建中国药包材标准体系, 推动药包材及药品行业的高质量发展。
来源:《医药导报》 2023 年7月第42 卷第7 期