您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-01-14 15:35
仿制药质量和疗效一致性评价启动五年!相关政策已经步入成熟,而一致性评价获批率却越来越低,为什么?对于与立项相关的问题又呈现出哪些新情况、新趋势?
焦点1 获批率越来越低,为什么?
一致性评价自2017 年开始受理,2019 年注射剂可以申报一致性评价,因此当年的一致性评价申报数量到达一个新的峰值,之后申报恢复到一个稳定数量。(详见表1)
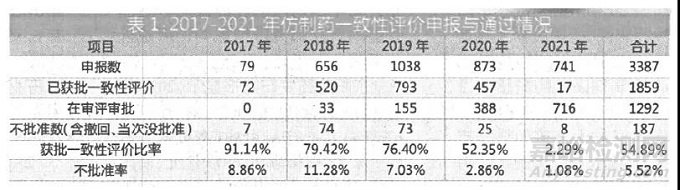
截至2021 年11 月15 日,2021 年申报一致性评价数为741 个,预计全年将与2020 年(873个)基本持平,难以超越2019 年的峰值(1038 个)。
通过率方面,2017 年申报一致性评价产品的获批率(即一致性评价通过率)为历年最高,超过90%。2018 年和2019 年一致性评价通过率都在70%~80%之间。
通过率方面,2017 年申报一致性评价产品的获批率(即一致性评价通过率)为历年最高,超过90%。2018 年和2019 年一致性评价通过率都在70%~80%之间。
焦点2 注射剂一致性评价
2018 年不批准率达峰值
2019 年9 月30 日,国家药监局综合司公开征求《化学药品注射剂仿制药质量和疗效一致性评价技术要求(征求意见稿)》《已上市化学药品注射剂仿制药质量和疗效一致性评价申报资料要求(征求意见稿)》意见;CDE 于2019 年11 月开始公开征求《化学药品注射剂仿制药(特殊注射剂)质量和疗效一致性评价技术要求(征求意见稿)》的意见。
2020 年5 月,随着国家药监局发布关于开展化学药品注射剂仿制药质量和疗效一致性评价工作的公告(2020 年第62 号),注射剂一致性评价才正式启动。
实际上,CDE 在2018 年就开始接受注射剂一致性评价的受理号,合计150 个,通过一致性评价101 个,不批准28 个,批准率67.33%,低于该年的批准率平均水平;不批准率18.67%,高于当年不批准率的平均水平。
2019 年注射剂的受理号430 个,通过一致性评价312 个,批准率72.56%,低于该年的批准率平均水平;不批准29 个,不批准率7.26%,低于当年不批准率的平均水平。
并非越早申报越好,为什么?
2018 年一致性评价受理号的不批准率达到历史峰值的原因还包括:2018 年不少企业申报了同一通用名注射剂的一致性评价,但该通用名注射剂的2018 年受理号都没有得以批准,直到2019年该通用名注射剂重新申报一致性评价后才获批准。
由此可见,对于注射剂一致性评价,并非越早申报越好。早期指导原则没有出台前,申报资料未必符合CDE 要求,等指导原则征求意见稿出台后申报,成功率会有所提升。
焦点3 处方变更影响几何?
大部分申报涉及工艺变更
根据《仿制药质量和疗效一致性评价受理审查指南(需一致性评价品种)》:未改变处方工艺的一致性评价申请或提出免于参加一致性评价的申请赋予CYHBXX(年份,两位)XXXXX 国(4字头,五位大流水)或JYHBXX(年份,两位)XXXXX 国(4 字头,五位大流水)的接收号;改变处方工艺的一致性评价赋予CYHBXX(年份,两位)XXXXX 国(5 字头,五位大流水)或JYHBXX(年份,两位)XXXXX 国(5 字头,五位大流水)的受理号作为处方工艺是否变更的识别。
而数据显示,大部分厂家申报一致性评价都发生了处方工艺变更,处方工艺变更与处方工艺未变更之间的比值高达9:1。
工艺未变更者,不批准率更高
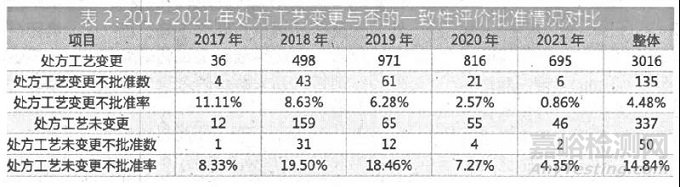
如表2 所示,2017 年没做处方变更的不批准率较低,主要是因为有不少产品当时是以新注册分类申报获批上市后再补充申请一致性评价的。2018-2021 年,处方工艺未变更的不批准率是处方工艺变更的3 倍左右。
由此可见,为了满足与原研药质量和疗效一致,即使原有的工艺发生了变更申请,也并不会影响获批的几率。
焦点4 大量将消逝的批文
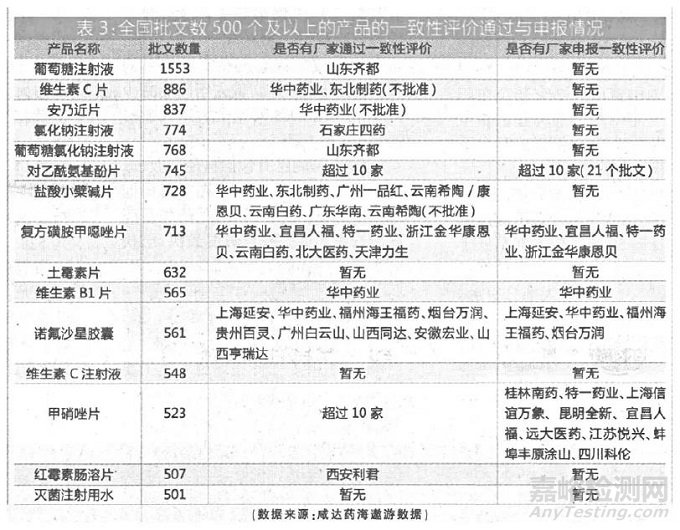
全国批文数500 个及以上的产品合计15 个,共有10841 个批文。咸达药海遨游发现,其中对乙酰氨基酚片、复方磺胺甲噁唑片、维生素B1 片、诺氟沙星胶囊和甲硝唑片已有厂家获批一致性评价,预计将有超过3000 个批文面临不能再注册。(详见表3)
焦点5 视同路径VS 非视同路径
存在“时间差”情况
通过一致性评价有两种情况:一是已上市的产品通过一致性评价研究获得通过一致性评价称号的路径(非视同路径);二是按新注册分类,获批后视同通过一致性评价(视同路径)。
理论上讲,两条路径对产品的审批要求是一样的,主要的不同是申报受理方式:前者是补充申请,后者是仿制药上市申请;前者已上市后者未上市,后者需要走一遍药品上市相关程序。
不过,当某种剂型的指导原则还没有出台的时候,一致性评价的申请(非视同路径)是不被受理的;但是,新注册分类的产品没有这个限制,于是就会出现“时间差”情况。比如,2017 年就有注射剂新产品获批上市并视同一致性评价(视同路径),而直到2018 年才开始有注射剂申报一致性评价(非视同路径),2020 年10 月才有第一个一致性评价获批(非视同路径)。
视同路径可能以较高价格集采中标
第一次国家集采的时候,就因为这个“时间差”,导致一众独家以新注册分类获批的注射剂厂家以较高价格在集采中标并抢夺已上市产品的市场。
即使目前集采的规则调整为只要通过一致性评价(含视同)和原研厂家在国内上市的数量达到3 家就可以启动国家集采,依然可能出现“时间差”情况:如果通过新注册分类路径通过一致性评价(视同路径)达到3 家,依然可以抢夺已上市厂家未过一致性评价的市场;而已上市厂家未通过一致性评价的,由于产品已经上市了,不能以新注册分类申报,也不能通过补充申请增加规格的方式获批上市并视同一致性评价。
两条路径可否统一“考核标准”?
如果一直以“没有相关剂型的指导原则出台”禁止企业申报一致性评价(非视同路径),那么对于已上市企业并不公平。无论已上市产品的一致性评价路径(非视同路径),还是以新注册分类申报仿制药视同通过一致性评价的路径(视同路径),最终都是要求仿制药与原研药质量和疗效一致。
因此,对于视同路径与非视同路径,有必要以统一的标准进行考核。对于已上市产品,注册程序可以考虑允许与新注册分类的受理和审评标准一致。比如,对于滴眼液、吸入剂等暂无剂型指导原则的一致性评价的剂型,已上市产品如果按照新注册仿制药分类的要求完成了一致性评价,是否也能得到仿制药一致性评价的批准呢?
此外,增加新规格的注册路径目前也不明确:企业应该走补充申请然后获批新规格视同一致性评价,还是走新注册分类获批视同一致性评价呢?

来源:医药经济报


