您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-05-10 08:16
研究背景
人无远虑,必有近忧。对于新药研发企业来说,何尝不想知道如何正确的制定研发策略,稳步推进项目向前发展,但是现实是新药开发之漫长与艰难,“百死千死一生”。有的时候,对于具体的项目,痛快的生,痛快的死,也是一种幸事。毕竟越往后花钱越多,越过二期的死亡之谷,三期仍然存在失败的可能性,甚至已经上市的产品,上市后因为开发不彻底等原因,很快退市仍旧多有发生。但是,面对高昂的回报,入局药物研发者仍然不绝于耳。
对于创新药研发,尤其是first in class创新药,每往前一步都是巨大的跨越。第一次苗头化合物的筛选,第一次先导化合物的筛选与优化,第一次确定了候选化合物,第一次开启了临床前研究,第一次申报IND,第一次进行临床实验......存在诸多第一次,同时每一个步调到下一个步调之间,还面临着诸多的决策。
对于制剂开发工作者,如何确定制剂处方开发的方向尤为重要。为了帮助科学家评估特定候选药物的口服吸收风险,已经开发了三个分类系统:生物药剂学分类系统(BCS)、可开发性分类系统(DCS)和精细可开发性分类系统(rDCS)。BCS作为监管指南,与早期评估候选药物在药物发现中口服给药的适用性以区分和选择临床候选药物的需要之间存在差距。
DCS于2010年发布,旨在缩小这一差距。DCS应用禁食状态模拟肠液中的溶解度来估计肠道溶解度,评估口服吸收过程中渗透性和溶解度的补偿性质,并提供了一种估计临界粒径的方法,在该方法下,溶出可能成为口服吸收的速率限制。
为了进一步实现候选药物和配方方法的务实比较,2018年推出了DCS的改进版本rDCS。rDCS是一种分两步进行的方法,在该方法中,典型分类系统中使用的标准研究首先进行,然后进行定制调查,以获得更深入的了解。这些研究通常基于确定与pH依赖性溶解度相关的溶出、过饱和和/或沉淀的影响,目的是确定克服不良或不一致吸收的配方策略。
总结三个分类系统的共同点不外乎都是基于药物的溶解性和渗透性为基点,评估药物分子体内吸收的风险,可见药物溶解性和渗透性这两个药物的基本理化性质对于药物研发的重要性作用。其实在前面推荐的几篇文章也无不涉及以上概念。
为什么在文章中去引入和强调各种分类系统,其实无论分类系统也好,决策树也罢,基本也是殊途同归,致力于通过处方前或者体外实验,对药物体内的吸收进行预测,一来评估后续制剂开发难以程度,二来找到后续制剂开发的风险点,三来对于后续制剂开发提供方向。此篇文章我们去了解一下强生的一些观点和思路。
强生制剂决策观点
根据生物药剂学分类系统,溶解度和渗透性是决定药物在胃肠道(GI)中被吸收能力的主要因素,从而影响口服生物利用度。渗透性较低的分子趋势可以通过产生具有更高分子量和/或增加氢键计数的先导化合物的药物发现趋势来解释。几种体外模型可用于评估发现阶段新候选药物的渗透性。Caco-2系统常用于评估新候选药物的渗透性,然后将结果与不同的FDA渗透性参考药物进行比较。渗透性问题可以通过使用不同的渗透性增强剂来克服,但它们的毒性通常是一个限制因素。由于配方技术不太可能解决渗透性差的问题,在制剂开发中不太容易开发生物利用度主要受渗透性限制的情况。
近年来,许多技术被开发出来,以克服新分子日益增加的溶解度问题。了解溶解度差的根本原因是选择最合适的溶解度的关键因素。在评估溶解度对口服生物利用度的影响时,应考虑重要的一点。事实上,溶解度和生物利用度之间的关系并不系统。这意味着我们可以看到一些水溶性差的分子,但仍然具有很高的生物利用度。这可以解释为胃肠道环境是一种动态介质,其中酶等生理因素和高渗透性的补偿影响足以纠正溶解度差。
这就引出了作者经常从他的药物化学同事那里收到的一个重要问题,当时正在讨论最近合成的难溶性分子:“我们能否通过增加水溶性来提高这种分子的生物利用度?真正的潜在问题应该是:“这种分子溶解度的生物利用度是否有限?为了简单回答这个问题,强生科学家建立了一个简单的决策树,用于处理生物利用度差的不同情况(图1)。
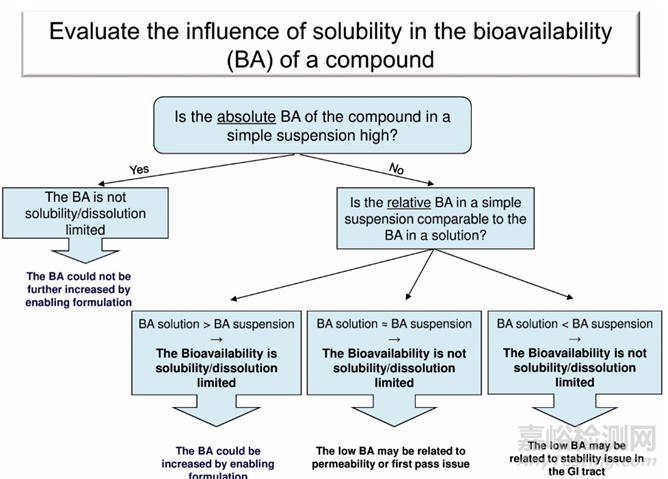
图1评估溶解度对化合物生物利用度(BA)的影响。2
该决策树在先导物优化过程中利用了通常可用的药代动力学(PK)研究信息。在开始一项漫长而昂贵的处方研究以提高溶解度之前,应明确确定口服生物利用度与特定化合物的溶解度之间的关系。进一步预测人类相对生物利用度(F相对),基于F相对大鼠数据,显示大致F相对大鼠>50%转化为F相对人类>90%。
图1决策树通过动物体内生物利用的研究,明确难溶性药物体内吸收的限速步骤:溶解性/溶出、首过效应还是因为药物在胃肠道的稳定性问题。
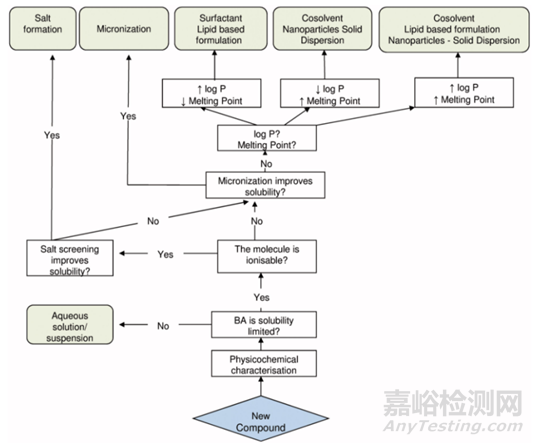
图2临床前制剂的决策树2
图2 临床前制剂的决策树作为决策树1的补充,很好的将判断难溶之因与解决办法进行衔接。在表征NCE之后,测试这是简单水溶液/混悬液制备的生物利用度是逻辑起点。为了验证生物利用度是否受溶解度限制,并可以获取分子的体内生物利用度数值。
决策树2从简单的溶液或混悬液制剂处方开始,如果需要提高溶解度,评估成盐的可能性是第一个推荐的方法(开发药物中绝大多数为可电离分子)。然后每当制备混悬液时,也强烈建议使用微粉化化合物。这是为了提高溶出速率,并有助于在未获得完全溶解时获得均匀的混悬液,如果问题未获得解决,再转向更复杂的技术,如API微粉化和潜溶剂/表面活性剂的使用。如果这些方法不能充分提高溶解度,则需要转向“新型配方”,例如纳米混悬液或固体分散体。应确定亲脂性和/或晶格力对溶解度差的贡献。
对于log P高、熔点低的分子,提高润湿性可增强吸收。这可以通过使用表面活性剂、脂质载体或脂质表面活性剂来实现。对于具有挑战性的分子,可以评估先进的脂质制剂,如自乳化药物递送系统(SEDDS)和自微乳化药物递送系统(SMEDDS)。
对于低log P和高熔点的分子,应首先评估在潜溶剂中的溶解度。由于溶质在胃肠道液中稀释后有以对数比例沉淀的风险,因此添加表面活性剂有助于将化合物保持为混悬液;如果溶解度的提高还不够,则需要纳米颗粒或固体分散剂来克服晶格力。
对于同时具有高熔点和高log P的分子,潜溶剂不允许充分暴露,应考虑固体分散体、脂质制剂和纳米颗粒等技术。
虽然没有普遍确定高log P和高熔点的明确分类,但在这个配方策略的范围内log P≥2-3和熔点≥160°C可以被认为是高的。很多资料中更多的认为log P在5/6以上,熔点在200℃以上,认为其数值处于高位。
开发案例
Telaprevir是一种在丙型肝炎病毒中发现的NS3:4a蛋白酶的小分子拟肽抑制剂。Telaprevir竞争性地作用于NS3:4a,通过以高亲和力结合蛋白酶的活性位点。仅基于结合亲和力,它作为抑制剂可能表现良好。然而,NS3:4a的活性位点主要是非极性的;因此,只有其他非极性化合物可能与它紧密结合。非极性-非极性相互作用的稳定性是由于它们的低相互作用自由能,无论机制如何,非极性靶点都是一个难题。非极性配体可能能够与靶位点紧密结合,但可能不高度溶于水或生理相关的水性介质中。Telaprevir具有高度疏水性,因此显示出非常低的水溶性。在水中,它的溶解度低于大理石。
Telaprevir化合物难溶的原因不是厌水,而是晶格的强度,由化合物LogP为2.8,熔点在246℃也可以得到确证。因此,破坏这种晶格可以增加整体溶解度。因此,一旦消除了作为障碍的晶格强度,Telaprevir的溶解度可以数量级提高,据图3也可以得出制备分散体,化合物体内吸收获得极大的提高。

图3 Telaprevir不同处方生物利用度统计4
参考文献
1.Current screening, design, and delivery approaches to address low permeability of chemically synthesized modalities in drug discovery and early clinical development
2.Rational formulation strategy from drug discovery profiling to human proof of concept
3.Leveraging the use of in vitro and computational methods to support the development of enabling oral drug products: An InPharma commentary
4.Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus

来源:药事纵横


