您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-04-24 11:02
“2024年5月26日前,遗留器械应当提交MDR符合性评估申请并建立质量管理体系,否则无法获得过渡期延长。”
1个月后条件不符的遗留器械不再享受过渡期延长!
欧盟发布的《MDR过渡期时间表》提到:
·仅限于2021年5月26日之前发布的公告机构证书或制造商的合格声明所涵盖的器械,才能从过渡期延长中受益。
·2024年5月26日,不符合新过渡期适用条件的遗留器械的过渡期结束。
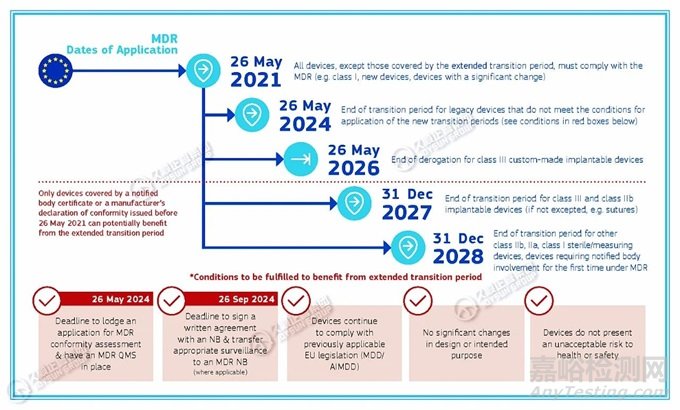
享受过渡期延长应当满足的条件有:
·2024年5月26日:正式提交MDR符合性评估申请并已建立MDR质量管理体系的截止日期;
·2024年9月26日:与公告机构签署书面协议并将适当的监督转移至MDR公告机构(如适用)的截止日期;
·器械继续遵守先前适用的欧盟法规(MDD/ AIMDD);
·设计或预期目的无重大变化;
·器械不会对健康或安全构成不可接受的风险。
哪些医疗器械属于遗留器械?
1. 在2021年5月26日前,MDD指令下的I类医疗器械已拟订符合性声明,MDR法规下其符合性评估程序需要公告机构参与的;
2. 在2021年5月26日前,按照MDD指令或AIMDD指令已取得CE标志的医疗器械。
MDR法规对质量管理体系有什么要求?
2024年5月26日前,所有遗留器械的质量体系建设必须符合MDR下EN ISO 13485:2016。
针对遗留器械,MDR法规新增许多质量体系要求:包括上市后监督、市场监督、警戒系统、经济运营商和器械登记等,对此制造商最晚不迟于2024年5月26日已建立质量管理体系。
MDR法规针对制造商质量体系引入不少新管理要求(包括以下内容):
·法规符合性策略(合规性评估程序、体系所涵盖器械重大变更的合规性等);
·确定适用的一般安全和性能要求GSPR,并探索符合前述要求的过程;
·管理层所承担的责任;
·资源管理(供应商和分包商的选择和控制);
·建立满足MDR\IVDR规定的风险管理过程;
·根据MDR法规要求开展的性能评估过程(包括:上市后性能跟踪PMPF);
·产品实现过程(包括:规划、设计、开发、生产和服务提供);
·根据MDR法规要求为器械分配UDI并确保所提供信息的一致性和有效性;
·根据MDR法规要求建立、实施和维护上市后监督系统PMS;
·与主管部门\公告机构\经济运营商\客户\其他利益相关者的沟通;
·在警戒背景下报告严重事件和现场安全纠正措施的流程;
·纠正和预防措施的管理及其有效性的验证;
·输出、数据分析和产品改进监控测量过程。

来源:久顺集团技术服务


