1、年度检查概况
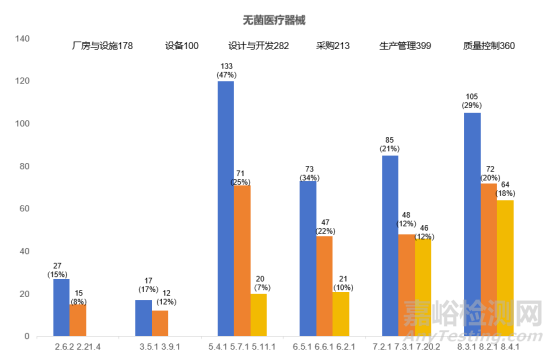
2023年度,本市无菌医疗器械现场核查合计发现缺陷1666项。厂房与设施(178项次)、设备(100项次)、设计开发(282项次)、采购(213项次)、生产管理(399项次)、质量控制(360项次)占比超九成,下面将对这六个方面的关键风险点及常见核查问题进行梳理分析。
2、现场核查常见问题
下面将从厂房和人员、设备、设计开发、采购控制、生产管理、质量控制四个方面对无菌医疗器械的常见核查问题进行梳理分析。
2.1 厂房和人员
2.6.2 仓储区应当按照待验、合格、不合格、退货或召回等进行有序、分区存放各类材料何产品,便于检查、和监控。
仓库分区标识不完整,例如未设置合格品/退货区;缺少货位卡或者货位卡的缺少关键信息;原材料包装缺少标签信息;试剂存储条件不符合要求,例如光照,温度不符合要求;不同物料混放。
2.21.4 在其他洁净室(区)内,水池或地漏应当有适当的设计和维护,并安装已于清洁且带有空气阻断功能的装置以防倒灌,同外部排水系统的链接方式应当能够防止微生物的侵入。
洁净车间密封不良,例如与普通车间相连的洁净车间门下方未做密封措施;洁净车间未设置下水防倒灌装置。
2.2 设备
3.2.1 生产设备的设计、选型、安装、维护和维修应当符合预定用途,便于操作、清洁和维护。
(1)设备未校准
未对设备进行校准,例如未对除湿干燥送料设备的温度参数进行校准;设备验证报告不完整,例如数控机床验证报告未包含运行验证和性能验证。
(2)设备故障
设备损坏,例如电脑平车(缝纫机)液晶屏破裂,无法正常显示。
3.2.3 应当建立生产设备使用、清洁、维护和维修操作规程,并保存相应的设备操作记录。
保养记录不完整,缺少日期或者人员签字;实际操作未按照保养规程操作;未见相关生产设备的保养规程。
3.5.1 应当配备适当的计量器具,计量器具的量程和精度应当满足使用要求,计量器具应当标明其校准有效期,保存相应记录。
未对计量器具进行校准或者部分关键参数进行校准,常见的情况有未对载荷试验机拉伸速度进行校准;无校准标识。
3.9.1 应当制定工艺用水的管理文件,工艺用水的储罐和输送管道应当满足产品要求,并定期清洗、消毒。
企业没有对外购纯化水的检验能力;纯化水系统使用维护保养标准操作规程未明确具体的日常保养方法,例如未规定对纯化水储罐进行消毒的频率。
2.3 设计开发
5.4.1 设计和开发输出应当满足输入要求,包括采购,生产和服务所需的相关信息、产品技术要求等。
1)产品图纸、采购质量标准等输出内容不完整。
无尺寸信息或尺寸信息不完整;当有多种规格时,仅部分规格有图纸。
2)产品技术要求、产品图纸、采购质量标准、成品检验指导书等技术文件之间存在不一致。
设计开发控制程序和项目建议书不一致;图纸信息不完整;图纸中的信息与技术要求不一致,常见的有编织方式,球囊壁厚,涂层位置、产品尺寸信息等;未提供提供总装图。
5.7.1 应当对设计和开发进行验证,以确保设计和开发输出满足输入的要求,并保持验证结果和任何必要措施的记录。
缺少老化验证报告;或者老化验证的时间与货架有效期不一致;或者老化验证项目不完整;缺少相关验证,例如未提供灭菌后产品性能的验证报。
5.11.1 应当在包括设计和开发在内的产品实现全过程中,制定风险管理的要求并形成文件,保持相关记录。
未提供风险评价报告,例如缺少产品不合格问题的风险分析报告;进货检验时间过久,未能就制定依据提供相应的风险评价。
2.4 采购
6.2.1 应当根据采购物品对产品的影响,确定对采购物品实行控制的方式程度。
采购清单不完整,未包含所有的采购物料,生产日期,销售出库时间。
6.4.1 应当与主要原材料商签订质量协议,明确双方所承担的质量责任。
缺少质量协议。
6.5.1 采购时应当明确采购信息,清洗表述采购要求,包括采购物品类别、验收准则,规格型号,规程、图样等内容。
产品技术要求和进货检验规程不一致;质量协议未明确采购要求,缺少质量标准。
6.6.1 应当对采购物品进行检验或者验证,需要进行生物学评价的材料,采购物品应当与经生物学评价的材料相同。
进货检验事项存在漏洞,例如进货检验项目不完整。
2.5 生产管理
7.2.2 应当编制生产工艺规程,作业指导书等,明确关键工序和特殊过程。
1)产品作业指导书缺少关键的生产参数。
2)产品作业指导书未明确规定物料的规格,牌号信息。
3)工艺流程图缺少关键工序,或未标明关键工序或特殊工序。
7.3.1 在生产过程中需要对原材料、中间品等进行清洁处理的,应当明确清洁方法和要求,并对清洁效果进行验证。
清洗最大量未验证,清洗温度要求未验证;进行清洗验证的组件缺乏典型性。
7.5.1 应当对生产的特殊过程进行确认,并保存记录,包括确认方案、确认方法、操作人员、结果评价、再确认等内容。
1)未提供相关设备的设备确认报告,例如缺少清洗设备的设备确认报告。
2)封口验证报告中封口的上下限验证不充分,封口设备未验证,封口机无对应的封口作业指导书中的参数,封口速度未验证或者与验证报告不一致。
3)生产过程中验证记录和验证报告不一致,生产相关参数的验证次数不符合文件要求。
7.6.2 生产记录应当包括:产品名称,型号规格,原材料批号、生产批号或产品编号、生产日期、数量、主要设备、工艺参数、操作人员等内容。
1)生产协议不完整,例如委托生产协议未明确生产批号的命名方式(含进货,过程和成品);委外灭菌协议未明确灭菌批号和生产批号的关系。
2)生产记录与生产技术文件不一致,如 包装封口参数不一致。
3)生产记录不完整,例如未记录设备编号,物料批号记录不完整,清洗时间、温度。
7.20.2 灭菌过程应当按照相关标准要求在初次实施前进行确认,必要时再确认,并保持灭菌过程确认记录。
1)灭菌确认报告不完整,例如灭菌再确认报告未记录短周期、全周期灭菌确认用样品的批次信息,也未提供相应的生产记录。
2)进行灭菌确认的产品型号不具有代表性。
3)灭菌记录未记录主要过程参数,例如预热温度、预热时间等。
2.6 质量控制
8.2.1 应当定期对检验仪器和设备进行校准或者检定,并予以标识。
仪器校准范围未包含该仪器的全部检测范围或者未覆盖是使用范围,例如拉力机的校准范围为50-200N,实际使用范围为1-10N。
8.3.1 应当根据强制性标准以及经注册或者备案的产品技术要求制定产品的检验规程,并出具相应的。
1)委外检测机构无相关检测的资质,或者化学性能等委外检测的项目未建立检验操作规程。
2)检验用水不符合检验规程要求。
8.4.2 检验记录应当包括进货检验、过程检验和成品检验的检验记录、检验报告或证书等。
检验记录不完整;实际检验方法和检验规程不一致;检测抽样数量不符合抽样规则要求。
3、总结建议
综合来看,无菌医疗器械现场检查的共性问题主要集中在生产管理,质量控制,设计开发、采购、厂房和人员及设备等方面。建议企业加强医疗器械相关法律法规的学习和考核,加强企业质量管理内审。监管部门也需进一步加强检查员的业务培训,重视监管手段的创新,共同维护医疗器械的质量可控和安全有效。