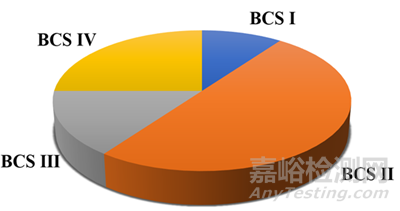
众所周知,因为口服制剂易于管理、稳定性好、生产工艺成熟、商品成本低,口服给药仍是药物给药途径的首要选择。对于口服给药药物递送策略来说,药物需要满足一定的口服生物利用度才能达到应有的药效,而药物在吸收部位溶解,将是决定药物吸收的重要因素。难溶性药物犹如雨后春笋般涌现,限制了药物在胃肠道吸收部位的溶解,限制了口服药物的吸收,最终影响了新药开发的体内暴露问题,可能造成项目正常的往前推进受到影响。一个比较严峻的现实是,已上市品种中大约75%药物为难溶性药物,而处于研发产线中难溶性的占比更加突出,甚至在90%以上(图1)。
图1 基于BCS分类系统已上市品种药物的分类占比[1]
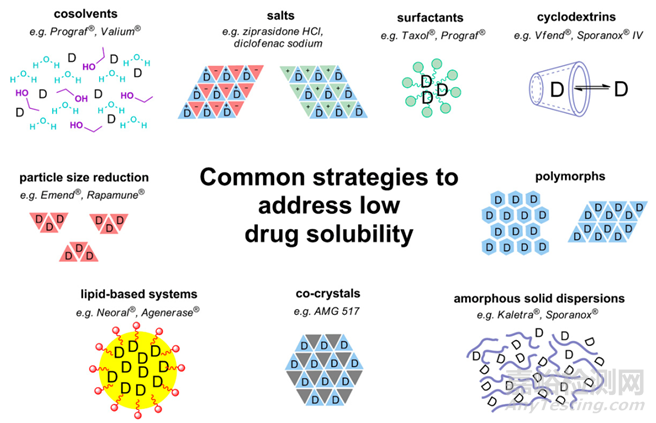
为了解决限制药物吸收的溶解度问题,提高药物的体内暴露,制剂科学家们探究了众多的增溶策略(图2),如原料粉碎,微粉甚至纳米化,环糊精包合技术以及在处方中加入助溶剂,潜溶剂,增溶剂,药物固态开发策略(包括多晶型筛选,盐型/共晶研究,无定形固体分散体制备)以及脂质制剂(如微乳,纳米乳等)。
图2 常见的难溶性药物增溶策略[2]
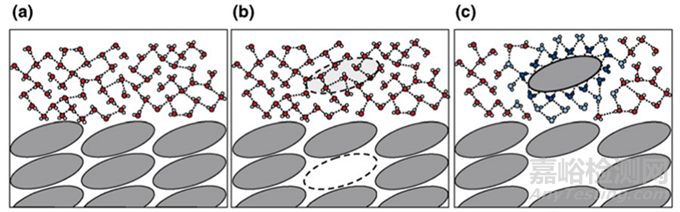
增溶策略众多,为了更好的有针对性的选择增溶策略,我们不妨从化合物的溶解过程去探究药物溶解的限速步骤。俗话说,知其然,知其所以然。了解化合物的溶解的原理(图3),针对其“症结”,“因病施治”,或许更容易“药到病除”。图3a描述了溶剂-溶剂,溶质-溶质分子间的分离状态以及其与其他类型的分子的相互作用;图3b描述了溶质分子转移到溶液的过程,首先溶质分子需要从晶格中脱离,即要打破晶格能,脱离溶质分子之间的作用力,然后溶剂分子要空出容纳溶质分子的空间,即离解溶剂分子之间的相互作用(空化能);图3c溶质分子进入溶剂后,溶剂分子与溶质分子将发生稳定的相互作用,这个过程需要的能量称之为溶剂化能(溶剂通常为水性溶剂)。
图3 化合物的溶解过程[3]
总结来说,一个晶体药物的溶解过程,涉及三种能量,即需要打破的晶格能,溶剂间空出空隙的空化能,溶质分子溶剂化的溶剂化能。一般认为,晶格能和溶剂化能能量要大于空化能,所以一个晶体化合物的溶解度较差通常认为与晶体药物溶解过程所涉及的晶格能和溶剂化能较强有关。
溶质分子间具有强的晶格能,限制了溶质分子的解离,这类化合物称之为“brickdust”,如石头般“顽固不化”。通常,使用化合物的熔点来显示其对于药物溶解度的限制,即熔点越高,晶格能越强,溶解度越低。熔点的临界值一般为200℃。
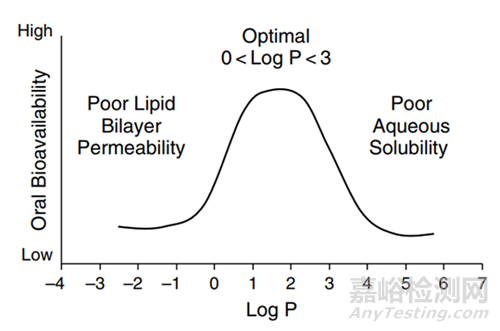
图4 LogP对于化合物口服生物利用度的影响[5]
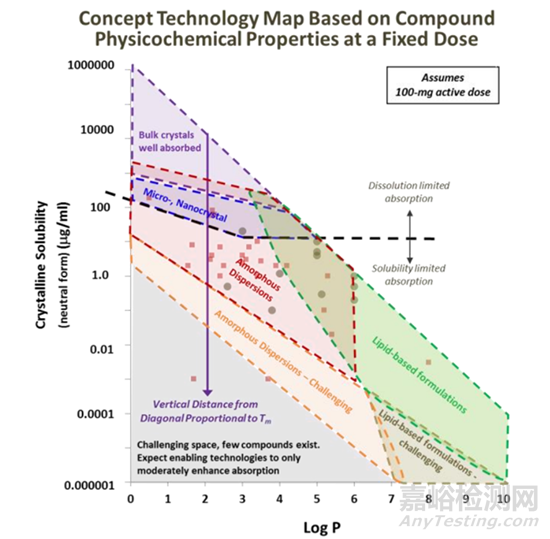
溶质分子需要与溶剂分子相互作用,“水乳交融”,但是一般疏水化合物与溶剂分子作用有限,此类高疏水性的难溶性药物称之为“greaseball”,通常描述水化作用的参数为油水分配系数LogP,有建议将LogP2-3作为溶剂化能是否会限制晶体化合物溶解的临界值,看更多的资料发现,很多科学家更会把LogP在5以上作为临界值(图4,5),Lonza对于logP值低于5,而熔点过高,晶格结构限制化合物溶解度的化合物,推荐采用固体分散体技术,使稳固的晶体结构土崩瓦解,呈现无定形状态;当LogP高于5,水合能力较差,疏水性强,可以通过脂质制剂,助力形成亚微米乳穿过未搅拌的水层,提高疏水药物体内吸收。一个基本的认识就是LogP数值越大,化合物疏水性越强。对于可电离的化合物分子,可能使用LogD会更加的准确。
图5 Lonza基本化合物的熔点和LogP作为增溶策略的选择依据[6]
比较现实的问题,有些化合物可能兼具熔点高和LogP大的特点,大概可以推测出此化合物可开发性极差,很难通过增溶的技术达到目标的生物利用度,当然,药物分子在胃肠道吸收的过程和机制极其的复杂,尚需进行全面的研究。从上可知,化合物结构导致药物分子用于口服给药具有挑战性的物理化学特性,从药物溶解度的角度提炼出了化合物两个最基本的理化性质参数熔点和LogP,可用于表征化合物的溶解度限制因素,从而可以有针对性的选择增溶策略。
举例:特拉匹韦具有高度结晶性,熔点246°C且具有比较强的疏水性,logP为4.0,导致结晶药物水溶性极差(4.7μg/ml),其溶解度实际上低于大理石(14μg/ml)。初步制剂研究,表明溶液、混悬剂甚至纳米晶混悬剂导致特拉匹韦的口服暴露量非常低。特拉匹韦配制成无定形喷雾干燥分散体,显著提高了在水性介质中的动力学溶解度,最终提高了生物利用度。
难溶性药物越来越多,溶解度的下限不断地刷新,正如特拉匹韦的案例所示,有些化合物的溶解度甚至要低于大理石。正如图5所示,对于熔点较高,化合物溶解度极差,仅仅通过固态形式的改变(粒度较小甚至纳米晶等手段)很难克服;对于一些酸碱性的化合物来说,成盐在一定程度上可以提高药物的饱和溶解度,但是如何筛选合适的反离子以及药物本身的酸碱性极弱,造成所筛选的盐型易于发生歧化反应,这个问题遇到的几率再增加,而且还有可能牺牲药物的其他的性质。
固体分散体增溶手段从概念的提出到上市产品的应用,已经走过了60年以上的时间,理论逐步的完善,实践在很多的品种上获得了成功。无定形固体分散体将稳定的晶态药物转变为高能的无定形形态,提高了难溶性药物的溶解度,是克服药物难溶性的有效手段之一。当然,在解决问题的同时,同样也伴随着新的问题的发生,例如物理稳定性问题。高能态的无定形形式势必要自发的转向低能的稳定晶型状态,犹如水往低处流淌一样,但是随着高分子材料的开发与应用,在维持无定形的物理稳定性上,虽然不能阻断转晶的发生,但是却可以延长转晶发生的时间,以保证药品的货架期。
总结:上文我们阐明了目前药物研发遇到的拦路虎-药物的难溶性,而且情况很是严峻。虽然增溶策略众多,但是如何进行快速的有效的有针对性的选择增溶策略呢?为了明晰这个问题,我们又简单介绍了一个晶体药物的溶解度过程,明确了可能限制溶解度的化合物理化性质-化合物的熔点及LogP,接着我们引用了Lonza内部的一个增溶策略决策图,证实了化合物熔点和LogP可用于指导选择增溶策略,而且对于极其难溶的药物很难通过一般粒度控制策略去解决,最后我们强调了无定形固体分散体在解决难溶性药物的机遇与挑战。本文更是像是一个前言或者引子,表明为什么会选择固体分散体技术以及它与其他增溶策略的不同点。
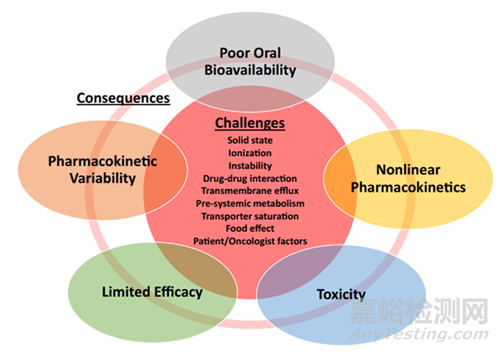
前文我们提到难溶性药物犹如洪水一般奔涌而来,药物无法溶解,将无法完成吸收,影响化合物的体内生物利用度,影响药物的疗效,除此之外还可能具有食物效应,造成病人体内PK变异性较大,甚至产生毒副作用(图6)。为了切实的解决难溶性药物问题,我们引入增溶策略-固体分散体技术。
图6 口服给药难溶性抗肿瘤药物随着带来的问题[1]
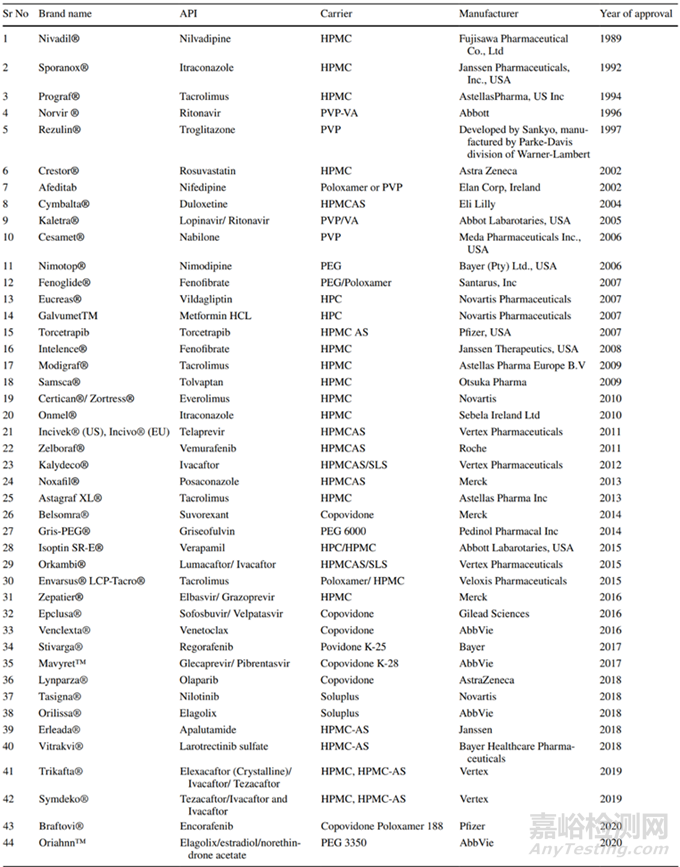
Sekiguchi和Obi于1961年首次报道了使用固体分散体作为提高水溶性药物溶出度和口服生物利用度的手段,制剂科学家们并在随后的60年中进行了广泛探索,目前已经应用固体分散体技术开发上市40个品种(图7)。
无定形态具有高能,溶解度要高于稳定晶型形式。制备的固体分散体中药物要么以相分离的结晶或无定形的形式混悬在聚合物材料中,要么以药物和聚合物的均匀分子混合物的形式存在。增溶的机制在很大程度上由所形成的固体分散体的结构决定,但在许多情况下,它是由相同材料的晶态和无定形态的溶解度和溶出行为的根本差异所支撑的。考虑到结晶和无定形药物溶解度通常存在很大差异,不同种类的固体分散体通常会导致溶出速率的显著差异。含有无定形形式药物的固体分散体通常比含有结晶药物的制剂表现出更快的溶出速率,从而显著改善药物吸收。
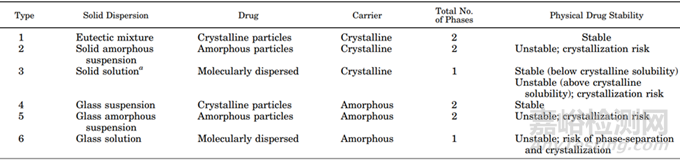
聚合物可以以无定形或结晶形式存在,其中聚合物分子还是以无定形存在居多。据文献报道固体分散体种类众多(图8),比较理想的状态是固体溶液,药物以分子的形式均匀分散在无定形的聚合物材料中;比较常见且易于制备的固体分散体更多的是药物以无定形形式分散体聚合物材料中,即无定形固体分散体。
图7 已上市以固体分散体技术为制备的产品[2]
图8 固体分散体的分类[3]
固体分散体通过多种机制增加药物溶出,包括有效粒径的减小、润湿性的改善、表面活性剂的增溶作用,通过将药物稳定在溶解度更高的无定形状态来消除晶格能的影响以及维持过饱和的时间。
俗话说,知其然,知其所以然!今天我们不妨去探究为什么在新的历史时期固体分散体成为难溶性药物众多增溶策略中的宠儿,它具有什么独特的功能以达到增溶的目的?
a.粒径减小。
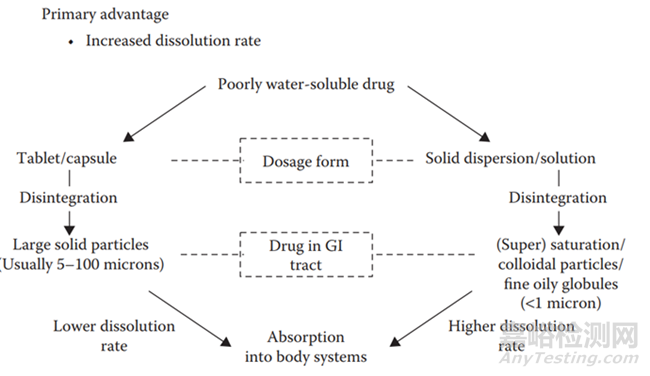
众所周知,原料的处理方式,比较常见的是锤式粉碎,一般原料的粒度可以粉碎到D90在10μm左右;如果想获得更低粒度的API,就不得不借助耗能更强的微粉,一般可以微粉到2-5μm,查文献接触到一个词,临界粒度,表明了一个道理,每一种粉碎技术对应一个所粉碎API粒度的下限;固体分散体中药物以混悬或分子分散在聚合物中的药物(无论是无定形的还是结晶的)。在大多数情况下,混悬的药物颗粒具有比传统固体制剂中通常更小的颗粒尺寸,并且在分子分散体的情况下,理论上,聚合物中药物的颗粒尺寸被最小化到单个分子的尺寸。颗粒尺寸的减小提供了溶出的有效表面积的增加。在一种比较极端的情况,制备的固体分散体物理不稳定,无定形转为了结晶型,可能制备的产品溶出依旧大于微粉处理的情况,因为固体分散体即使析晶所产生的聚集体的粒度可能都要小于2μm。根据经典的Noyes-Whitney方程,比表面积的增加,药物溶出速率增加,进而可以提高药物吸收部位的药物浓度,提高药物吸收与暴露,最终提高药物的生物利用度。与传统制剂比较,固体分散体通过降低了API粒度,提高其比表面积,进而增加了药物的溶出速率(图9)。
图9 与传统的片剂或者胶囊制剂相比,开发的固体分散体在提高药物溶出速率和生物利用度方面上的优势。[4]
b.药物润湿性和增溶。
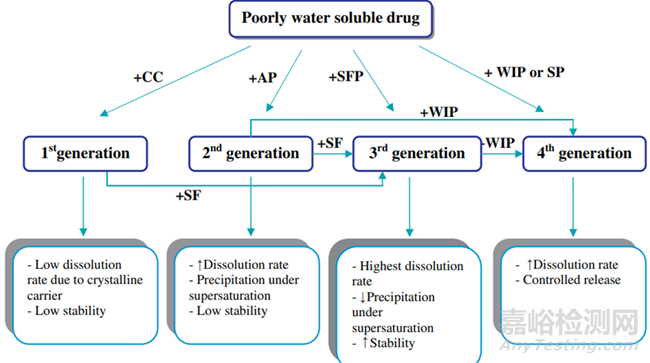
高水溶性聚合物材料的使用还促进水性介质快速流入固体分散体,从而增加润湿性。为了利用这种效果,第一代固体分散体使用低分子量、高度水溶性的聚合物,例如尿素,甘露醇,山梨醇等。在晶体固体分散体中,晶体药物分散在晶体聚合物内,形成共晶或单晶混合物。增加药物溶解度和溶出速率的主要方法有减小粒径、提高润湿性和改变多晶型。
图10 开发的固体分散体所用的聚合物材料的迭代发展[5]
聚乙二醇(PEG)是第一种被广泛开发的第二代聚合物辅料,是第一个商业固体分散产品所采用的聚合物。PEG可以通过热熔挤出容易地与药物融合,然而,与药物和聚合物的简单物理混合物相比的益处并不总是显而易见的。在无定形固体溶液中,药物和无定形聚合物可以完全混溶,形成分子均质的混合物,而无定形固体混悬液是由两个独立的相组成。第二代固体分散体主要包含无定形聚合物聚合物,无定形聚合物主要为全合成聚合物包括聚维酮(PVP),聚乙二醇(PEG),交聚维酮(PVP-CL),聚乙烯吡咯烷酮-共乙酸乙烯酯(PVPVA)和聚甲基丙烯酸酯。天然产物的聚合物由纤维素衍生物组成,例如羟丙基甲基纤维素(HPMC),羟丙基纤维素(HPC),羟丙基甲基纤维素邻苯二甲酸酯(HPMCP),羟丙基甲基纤维素乙酸琥珀酸酯(HPMCAS)等。
API既可溶解并混悬在聚合物中,也可同时以无定形和结晶状态存在。在无定形固体分散体中,API的尺寸非常小(分子,无定形颗粒或小晶体),而以超饱和状态存在于无定形聚合物中。当无定形固体分散体溶于水时,无定形聚合物可以提高药物的润湿性和分散性,并抑制药物的沉淀过程。由于无定形聚合物的热力学稳定性低,溶出速率快,增强了药物的溶解度和释放速率。
构建第三代固体分散体在聚合物包括表面活性,可促进制剂的乳化和药物的增溶。许多表面活性剂已被添加入固体分散体中,包括离子型(如十二烷基硫酸钠)和非离子型表面活性剂(如Pluronic嵌段共聚物)。具有两亲结构的表面活性剂可增强药物和聚合物的混溶性,从而降低药物的重结晶速率。而且,表面活性剂或乳化剂通过吸收到药物颗粒的外层或形成胶束来封装药物,从而提高药物的润湿性并防止由于过饱和而引起药物沉淀。第四代固体分散体是控释固体分散体,其主要应用于水溶性差,生物半衰期短的药物。
总结:固体分散体增溶策略充满着机遇以及挑战。但是,我们仍然清楚看到其无限的应用前景和生命力。目前已经上市固体分散体品种中,以喷雾干燥和热熔挤出工艺在固体分散体开发中应用最为广泛,工艺的选择和工艺参数的设定都会直接影响所制备的固体分散体的性质,如固体分散体粒度及其粒度分布,物理稳定性,结晶度等。这些固体分散体的关键理化性质又会进一步影响药物在体内外的溶解度和溶出的情况,最终会对体内的生物利用度造成影响。固体分散体的稳定性需要聚合物来提供相互作用力,以保证所制备的固体分散体在体内外水性环境过饱和时间的维持以及或货架期的储藏环境的物理稳定性(保持无定形状态)。当然,无论是固体分散体的粒度,药物在聚合物中分布状态及形式以及聚合物材料的种类及处方组成,都在一定程度上助力于难溶性药物溶解度的提高。后续文章,我们将进一步介绍固体分散体增溶的比较核心的机理。
参考文献
[1] Di L , Kerns E , Carter G .Drug-Like Property Concepts in Pharmaceutical Design[J].Current Pharmaceutical Design, 2009.DOI:10.2174/138161209788682479.
[2] Patel K , Shah S , Patel J .Solid dispersion technology as a formulation strategy for the fabrication of modified release dosage forms: A comprehensive review[J].Daru : journal of Faculty of Pharmacy, Tehran University of Medical Sciences, 2022, 30(1):165-189.DOI:10.1007/s40199-022-00440-0.
[3] Bhattachar S N , Deschenes L A , Wesley J A .Solubility: it's not just for physical chemists[J].Drug Discovery Today, 2006, 11(21-22):1012-1018.DOI:10.1016/j.drudis.2006.09.002.
[4] Bergstrom,Christel,A,et al.Computational prediction of drug solubility in water-based systems: Qualitative and quantitative approaches used in the current drug discovery and development setting[J].International Journal of Pharmaceutics, 2018.
[5] Kerns E H , Di L .Drug-Like Properties: Concepts, Structure Design and Methods[M].Elsevier LTD, Oxford,2008.
[6].Lonza:Technology Selection for Bioavailability Enhancement(https://www.lonza.com/knowledge-center/smallmolecules/es/oral-bioavailability-enhancement-programs)
[7] Anane-Adjei A B , Jacobs E , Nash S C ,et al.Amorphous solid dispersions: Utilization and challenges in preclinical drug development within AstraZeneca[J].International journal of pharmaceutics, 2022, 614:121387.DOI:10.1016/j.ijpharm.2021.121387.
[8] Kwong A D , Kauffman R S , Hurter P ,et al.Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus.[J].Nature Biotechnology, 2011, 29(11):993-1003.DOI:10.1038/nbt.2020.
[9] Vasanthavada M , Tong W , Serajuddin A T M .Development of Solid Dispersion for Poorly Water-Soluble Drugs[M]. 2008.
[10]Williams, Hywel D ,Trevaskis,et al.Strategies to Address Low Drug Solubility in Discovery and Development[J].[2023-11-13].
[1] Gala U H , Miller D A , Williams R O .Harnessing the therapeutic potential of anticancer drugs through amorphous solid dispersions[J].Biochim Biophys Acta Rev Cancer, 2020(1).DOI:10.1016/j.bbcan.2019.188319.
[2] Patel K , Shah S , Patel J .Solid dispersion technology as a formulation strategy for the fabrication of modified release dosage forms: A comprehensive review[J].Daru : journal of Faculty of Pharmacy, Tehran University of Medical Sciences, 2022, 30(1):165-189.DOI:10.1007/s40199-022-00440-0.3..Development of Solid Dispersion for Poorly Water-Soluble Drugs
[3] Williams, Hywel D ,Trevaskis,et al.Strategies to Address Low Drug Solubility in Discovery and Development[J].[2023-11-13].
[4] Liu, R. (Ed.). (2017). Water-Insoluble Drug Formulation (3rd ed.). CRC Press. https://doi.org/10.1201/9781315120492
[5] Vo L N , Park C , Lee B J .Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs[J].European Journal of Pharmaceutics & Biopharmaceutics, 2013, 85(3):799-813.DOI:10.1016/j.ejpb.2013.09.007.