口服给药是首选的给药途径,口服药物的生物利用度在药物发现阶段先导物选择中起着重要作用,并且是许多新药开发决策的关键。口服生物利用度取决于许多因素,主要包括药物渗透性、溶解性、溶出率、首过代谢和外排转运等。在这些因素中,低渗透性和低溶解度是导致口服生物利用度低的最常见原因。
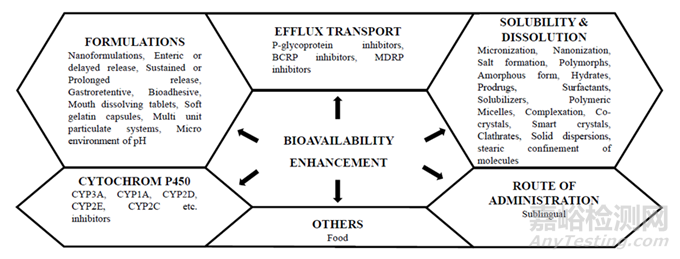
1997年,Lipinski等提出了药物的“5法则”,该法则成为预估口服药物吸收的参考。因此,根据此原则也提出了许多预测口服生物利用度的方法。预测方法主要关注药物特性,即通过脂质膜的溶解度和渗透性。对大部分药物来说是有用的,但对于通过转运蛋白吸收的药物,如β-内酰胺类抗生素、血管紧张素转换酶抑制剂和核苷类抗病毒药物,往往无法达到理想效果。此外,早期预测生物利用度的方法还忽略了一些因素,如潜在的高系统性前和/或首过代谢以及对外排泵的易感性,包括p -糖蛋白(Pgp)或多重耐药蛋白(MRPs),这些因素对某些类型的药物生物利用度有重大影响。为提高口服生物利用度而制定的策略如图1所示,通常组合使用。充分了解药物低生物利用度的原因和首选吸收途径,以及不同途径对药物代谢和药代动力学特征的影响,从而制定提高药物生物利用度的有效策略,是早期成功选择特定候选药物的关键。
图1 提高口服药物生物利用度的思路
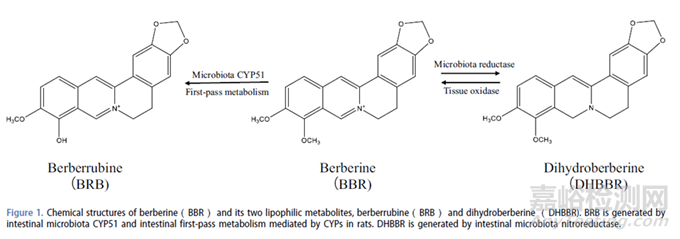
黄连素(Berberine,BBR)是一种季铵异喹啉类生物碱,具有许多药理活性,结构如图2所示。BBR在大鼠和人类中的口服生物利用度相当低,因此BBR的临床应用受到限制。在生物制药分类系统(BCS)中,BBR被归类为高溶解度和低渗透性的BCS III类药物。BBR是一种具有低亲脂性的阳离子离子化化合物。BBR的溶解度低,特别是在酸性条件下易形成盐,如BBR氯化物。此外,BBR与各种有机阴离子形成亲脂性离子对络合物。BBR是ATP依赖性外排转运体、P-糖蛋白(P-gp)和细胞色素P450(CYPs)的底物,在小肠和肝脏中大量表达。
图2 BBR及其两种亲脂性代谢产物BRB和DHBBR的化学结构
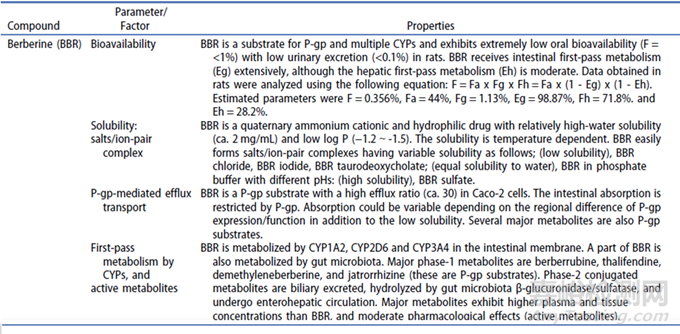
大鼠静脉(iv)输注15分钟,门静脉内(portal)输注15分钟,胃内(ig)或十二指肠内(id)给药BBR。BBR的剂量和血浆AUC0-36h分别为:ig,100mg/kg,57.9ng·h/mL;id,100 mg/kg,59.3 ng·h/mL;portal,4 mg/kg,466.0 ng·h/mL;iv,4 mg/kg,649.2 ng·h/mL。未被吸收的BBR部分占总剂量的56%,其中32%从大鼠粪便中检出,24%在口服(胃内)给药36小时后从整个胃肠道中检出,表明Fa(the fraction absorbed by the intestinal membrane,Fa)为44%。使用口服和静脉注射数据估计的绝对口服生物利用度(F)为剂量的0.356%。Fh的值估计为Fh=4 mg/kg剂量下的AUCportal /AUCiv,Eh的值估算为Eh=1-Fh,分别为71.8%和28.2%。通过F除以(Fa x Fh)估计的Fg值为1.13%,表明Eg=98.87%。这意味着口服给药后门静脉中完整的BBR(Fa x Fg=0.497%的剂量),表明通过在大鼠中肝首过代谢(Eh=28.2%)而进一步降低到0.356%。与AUCig和AUCid相比,在从胃向十二指肠的过渡过程中,BBR略有降解(约为剂量的2.4%)。
在10、20、40和60分钟时,大鼠原位空肠环中BBR的Fa吸收率分别为19.1%、26.5%、26.8%和33.6%。口服BBR(200 mg/kg)的大鼠在48小时内的尿液排泄率为BBR剂量的0.0939%,在24小时内的胆汁排泄率为9.2×10−6%,在48小时后的粪便排泄率为22.74%。此外,当大鼠口服BBR(48.2 mg/kg)时,BBR 84小时的累积尿排泄率为0.0101%,36小时的胆汁排泄率为0.174%,84小时的排泄率为剂量的8.43%。
BBR的绝对口服生物利用度非常低,低于剂量的1%,主要是由于肠首过代谢高(Eg,超过吸收量的98%)、肠吸收不足(Fa,约为剂量的50%)和肝首过代谢(Eh,其中20-30%到达门静脉)。BBR的肠道吸收不足是由于低溶解度和P-糖蛋白(P-gp)介导的外排转运。BBR与各种阴离子盐形成亲脂性盐/离子对络合物,并且络合物的溶解度根据反离子而变化。此外,BBR可引起P-糖蛋白介导的药物-药物相互作用。肠道CYPs介导的首过代谢是BBR口服生物利用度的主要降低因素。许多活性代谢产物是在肠道首过代谢后产生的。黄连素胆汁分泌的II期代谢产物被微生物群β-葡萄糖醛酸酶/硫酸酯酶水解并进行肠肝循环。肠道微生物群介导的代谢产生亲脂性代谢产物,包括具有更高分布体积的活性代谢产物小檗红(BRB)和二氢黄连素(DHBBR)。BRB在肠/肝中代谢为葡萄糖醛酸结合物,进行肠肝循环并发挥药理活性。BBR药代动力学特性如图3所示。
图3 BBR药代动力学特性
大鼠体内BBR生物利用度数据的药代动力学分析显示,口服生物利用度受到广泛的肠道首过代谢、低溶解度和P-gp介导的外排转运导致的膜通透性不足以及肝脏首过代谢的限制。根据理化性质(低溶解度、盐/离子对复合物的形成)和药代动力学性质(P-gp/CYPs的底物、广泛的肠道首过代谢),开发了各种剂量制剂来提高BBR的口服生物利用度,如图4所示。
图4 提高BBR口服生物利用度的制剂
配方增加了溶解速率/溶解度,有P-gp抑制剂的制剂,有表现出P-gp和/或CYPs抑制剂的增溶剂的制剂,有吸收促进剂的制剂,有胃/十二指肠滞留制剂,脂质制剂,针对淋巴转运的制剂,增加亲脂性的物理化学修饰。在这些制剂中,可以减少肠道首过代谢的制剂,如含有对P-gp和CYPs具有抑制作用的增溶剂的配方和含有吸收促进剂如C10、SDC或SNAC的配方,显著提高了BBR的口服生物利用度。确定哪个器官(肠或肝)负责BBR的广泛代谢;研究可以避免首次通过代谢的其他给药途径,如口腔、鼻腔、直肠和舌下给药,对于提高BBR的生物利用度也很重要。利用肠道菌群介导的亲脂性代谢物,如具有高膜通透性和更大分布体积的活性代谢物BRB和具有低P-gp外排比的BBR前体DHBBR,可以有效地分布到大脑,这是增强BBR体内药理活性的另一个重要思路。
参考资料:
1. DOI: 10.1517/17425247.2.3.419
2. DOI: 10.2174/1574892809666140917103834
3. DOI: 10.1080/17425255.2023.2203857
4. DOI: 10.1080/17425255.2023.2203858