摘 要 Abstract
为鼓励创新,加快新药研发,国家药品监督管理局药品审评中心组织起草制定了《化学药品创新药Ⅲ期临床试验前会议药学共性问题及相关技术要求(试行)》,并正式发布。本文结合国内创新药Ⅲ期临床试验前药学方面沟通会议审评中发现的问题,对Ⅲ期临床试验前药学共性问题进行分析总结,帮助申请人更好地理解并运用该技术要求,提高创新药药学沟通交流的质量和效率。
To encourage innovation and accelerate new drug development, the Center for Drug Evaluation has initiated the drafting and official release of the Pharmaceutical Common Issues and Related Technical Requirements in Pre-phase III Clinical Trial Meetings for Innovative Chemical Drugs (Trial). Drawing from issues identified in the review of pre-phase Ⅲ clinical trial meetings of pharmaceutical communication for innovative drugs in China, this paper analyzes and summarizes the recurring pharmaceutical common issues in such trials. The aim is to enhance applicants’ understanding and utilization of these technical requirements, thereby improving the quality and efficiency of pharmaceutical communication concerning innovative drugs.
关键词 Key words
创新药;Ⅲ期临床试验;药学共性问题;技术要求
innovative drugs; phase III clinical trial; pharmaceutical common issues; technical requirements
2020 年7 月1 日实施的《药品注册管理办法》明确指出,申请人在药物临床试验的关键阶段,可以就重大问题与国家药品监督管理局药品审评中心(以下简称药审中心)进行沟通交流[1]。为加强对药物研发与技术审评沟通交流工作的管理,药审中心于2020 年12 月发布了《药物研发与技术审评沟通交流管理办法》[2],进一步明确了沟通交流的分类及相关要求。
为鼓励新药研发和注册申报,加快新药上市进程,药审中心先后发布了《化学药品创新药Ⅰ期临床试验申请药学共性问题相关技术要求》(2020 年11 月)[3]、《创新药(化学药)临床试验期间药学变更技术指导原则(试行)》(2021 年3 月)[4]、《化学药品创新药上市申请前会议药学共性问题及相关技术要求》(2021 年11月)[5] 等一系列创新药药学研究相关指南。
随着上述技术要求和指导原则的发布,业界对Ⅲ期临床试验前沟通交流会议相关技术要求的呼声越来越高。为鼓励创新,加快创新药的研发进程,药审中心结合国内外法律法规、指导原则等,组织起草了《化学药品创新药Ⅲ期临床试验前会议药学共性问题及相关技术要求( 试行)》(以下简称《技术要求》)[6],并于2023 年3 月22 日正式发布。本文结合国内创新药Ⅲ期临床试验前药学沟通会议( 以下简称Pre- Ⅲ期药学会议)审评中发现的问题,对Ⅲ期临床试验前药学共性问题进行分析总结,帮助申请人更好地理解并运用《技术要求》,提高创新药药学沟通交流的质量和效率。
1、 创新药沟通交流会议召开情况及发现的问题
自2015 年药品审评审批制度改革以来,国家药品监管部门批准了多个化学药品创新药上市,提高了用药可及性,满足了临床用药需求。创新药的临床期间沟通交流在药物研发过程中发挥了重要作用,申请人与审评团队就重大技术问题及时沟通,有助于加快新药研发上市。
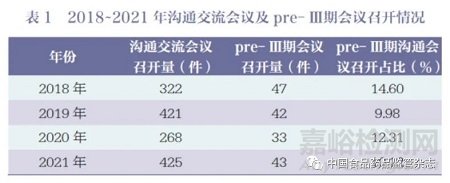
《2021 年度药品审评报告》指出,2021 年药审中心接收沟通交流会议申请4450 件,同比增长37.81% ;办理沟通交流会议申请3946 件, 同比增长61.00%。在药物研发关键阶段召开的Ⅱ类会议占比69.23%,其中,新药临床前(Pre-IND)申请办理占比32.84%,新药生产前(Pre-NDA) 申请办理占比11.05%,完成Ⅱ期临床后(即pre- Ⅲ期沟通交流)申请办理量为308 件, 办理占比7.81%[7]。2018~2021 年沟通交流会议及pre- Ⅲ期会议召开情况见表1。
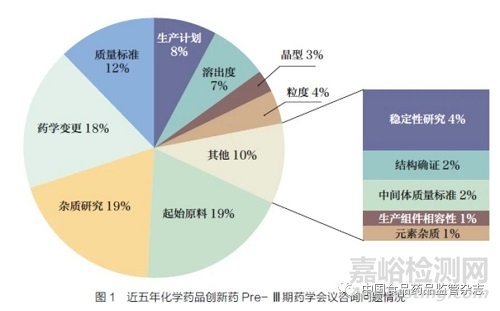
在《技术要求》起草初期,起草小组调研了近五年审评部门承办的42 个国内化学药品创新药Pre- Ⅲ期药学会议召开情况,包括召开面对面沟通交流、视频会议、电话会议及书面答复。这些案例中,包含179 个咨询问题,除去政策法规方面的24 个问题,其他155 个问题中,讨论频率较高的问题包括起始原料选择、杂质研究、药学变更等。其他问题包括稳定性研究、结构确证、中间体质量标准、生产组件相容性等,占总问题的10%,见图1。调研过程中,起草小组发现,随着《创新药(化学药)临床试验期间药学变更技术指导原则(试行)》的发布实施,2021~2022年的Pre- Ⅲ期药学会议中关于药学变更的沟通问题呈下降趋势。
调研发现,Pre- Ⅲ期药学会议中存在如下问题:①申请人对Pre- Ⅲ期药学会议重视程度不够,未提出药学沟通申请。②Ⅲ期临床试验前或Ⅲ期临床试验阶段亟待解决的问题,如起始原料的选择,直至完成Ⅲ期临床试验后仍悬而未决,影响药品上市申请进度。③药学研究深度不够,开发计划前瞻性不够,Ⅲ期临床试验期间仍然有重大药学变更,影响Ⅲ期临床试验结果的科学性。④提出的药学沟通内容不具体,提交的支持性研究资料不充分,影响沟通交流质量。
我国医药产业升级进程不断加快,新技术、新方法、新标准加速迭代,创新药申报数量的急剧增加,药物从研发到上市的进程加快,良好的沟通交流愈发显得重要。沟通交流作为审评团队与申请人的重要沟通渠道,有助于加快药物研发上市。
2、 Pre-Ⅲ期药学沟通交流的总体考虑
创新药Ⅲ期临床试验是用于支持上市的核心安全有效性证据的确证性临床试验,是药物的关键临床试验。Ⅲ期临床试验阶段也是积累工艺认知,加深对产品理解,制定商业化生产控制策略的重要阶段。创新药Pre- Ⅲ期药学会议为创新药研究期间的重要药学沟通交流会议,主要目的是解决关键临床试验开展前药学方面的重大技术问题。
《技术要求》作为创新药研发及注册期间沟通交流技术要求的一部分,主要讨论药学共性问题及相关技术要求, 为创新药Pre- Ⅲ期药学会议提供指导。《技术要求》适用于化学创新药和改良型新药。
申请人为创新药研发和注册申报的责任主体,对药物研发和注册申报负主体责任。一方面,基于药物自身特点及创新药阶段性和渐进性特点,结合药物临床试验进展情况,合理制定药学开发策略和开发计划。另一方面,申请人作为主体责任人,对品种的认识是最深入的,在开展沟通交流前,全面梳理药学研究结果,有助于查漏补缺。开展Ⅲ期临床试验前,药学研究需满足《创新药(化学药)Ⅲ期临床试验药学研究信息指南》的相关要求。《技术要求》在向社会征求意见时,业界对准备详细的研究资料和研究数据提出了较多疑问,需要说明的是,为保证沟通交流的质量及效率,建议申请人准备详细的研究资料和研究数据,资料越详实,越有助于解决申请人提出的问题。另外,建议提出的问题应尽量具体、有针对性。
在Pre- Ⅲ 期药学会议中,涉及工艺复杂的原料药或复杂制剂的沟通问题较多,情况也比较复杂,除《技术要求》中提及的问题外,还包括工艺控制、临床期间的质量桥接研究等。为保证Ⅲ期临床试验用样品的质量,支持Ⅲ期临床试验的开展及上市申请,需要开展更多的研究工作。基于上述考虑,建议这一类复杂品种在Ⅲ期临床试验前一定要与监管机构进行沟通。
通常,附条件批准上市药品的药学、药理毒理学要求与常规批准上市药品相同[8]。对于临床研究进展较快,预期临床有明显获益,拟采用药品加快上市注册程序(如附条件批准程序等)的创新药,或者临床开发周期比较短的创新药,申请人需尽早开展药学研究,药学研究计划需与临床研究计划相匹配,药学研究进度不应成为影响药物上市的限速步骤。
3、 常见的药学共性问题及审评考虑
3.1 起始原料选择
起始原料选择是Pre- Ⅲ期药学会议中非常重要的讨论问题。审评中发现,有些申请人并未重视这个问题,导致在上市申请前沟通交流或上市申请时要求前延合成路线,影响注册核查工作的开展,进而影响药品上市进程。
如果产品计划采用药品加快上市注册程序(如附条件批准程序等),需在用于支持上市的关键Ⅱ期临床试验开展前与审评机构讨论起始原料的选择问题。
审评工作中发现,起始原料的选择经常会遇到如下问题:①拟定起始原料选择依据不充分。②提供的杂质分析不全面。③起始原料中引入的杂质在后续工艺步骤汇总的清除转化研究不全面,无法支持目前起始原料的选择。
起始原料的选择需按照ICHQ11[9] 及问答文件[10] 提出的基本原则开展研究:①起始原料应当具备明确的化学特性和结构,未分离的中间体通常不被考虑作为合适的起始原料。②起始原料作为重要的结构片段并入原料药的结构中。③通用技术文件(CTD)“3. 2. S. 2. 2”应包含对原料药的杂质谱产生影响的生产步骤。④应充分描述原料药的生产工艺,明确杂质在工艺过程中的产生、去向与清除,控制策略满足杂质控制要求。⑤采用汇聚型原料药生产工艺的每个分支开始于一个或多个起始原料。每个分支从首次使用起始原料的步骤开始,需要遵循生产质量管理规范,并结合适当的控制策略为原料药的质量提供保证。⑥在生产工艺开始阶段附近发生的物料属性或操作条件的改变对原料药质量的潜在影响较小。需要特别说明的是,在选择起始原料时应当考虑上述全部原则,而不是孤立地严格遵循每一个原则。
在论证起始原料选择合理性时,可以提供包括但不限于以下资料作为支持性依据:①起始原料的杂质谱分析是否全面,分析方法检测起始原料中杂质的可行性,是否经过适当验证。②杂质及其衍生物在后续工艺中的去向和清除研究情况。③起始原料的质量标准如何实现整体的质量控制策略,可以结合中间体杂质研究情况论证起始原料的质量控制对终产品控制策略的影响。
ICH Q11 及问答文件阐述了半合成原料药的起始原料选择的相关考虑。通常情况下,应从源物质(微生物或植物)开始描述生产工艺。考虑到发酵/ 提取工艺及纯化工艺比较复杂,产品组分比例和杂质水平受工艺影响较大,建议将微生物发酵/ 提取步骤纳入原料药生产工艺,并将微生物发酵/ 提取及分离纯化工艺纳入原料药生产质量管理体系。ICHQ11 指出,如果能够证明合成工艺中的一个分离中间体符合上述化学合成原料药起始原料的选择原则,则该分离中间体可被提议作为起始原料。此种情况,建议申请人尽早与监管机构进行讨论。
在审评中发现,申请人由于原料供应等问题,会选择两家或多家起始原料生产商,但相关研究并不充分,不能证明不同生产商的起始原料具有可替代性。不同来源的起始原料的研究可参考《已上市化学药品药学变更研究技术指导原则(试行)》[11] 的相关要求开展。根据生产商的制备工艺,并结合杂质的清除转化研究情况,合理制定起始原料的质量控制策略。
3.2 有关物质
创新药的研发具有阶段性和渐进性的特点,随着临床试验的进展,对药物特性(原料药的合成工艺及降解途径)的认知不断加深,原料药及制剂的杂质谱分析会逐渐完善。如何保证有关物质分析方法能有效分离和检出潜在杂质?通常情况下,有关物质分析方法的开发是在杂质谱分析全面的基础上,结合化合物及杂质的理化性质,采用不同分离原理的分析方法,如高效液相色谱法、气相色谱法、离子交换色谱法、分子排阻色谱法等,进一步证明方法对潜在杂质的分离和检出能力;对于采用液相色谱法,需对色谱柱、检测波长、洗脱程序等进行筛选;用于方法开发的样品需具有一定的代表性,结合产品特性,可以从定向制备的杂质、含一定杂质的粗品或粗品母液(如5%~10% 杂质含量)、适度降解的样品、加速和长期稳定性样品等样品中进行选择。如果制剂工艺中用到热熔挤出、高温灭菌条件(如121℃、15min 灭菌)等工艺,需考虑有关物质方法能否对该苛刻条件下产生的潜在降解杂质具有较好的分离和检出能力。
有关物质是药物的关键质量属性。临床试验期间,申请人需不断积累杂质的检测数据。如果有关物质分析方法发生变更,需关注不同方法间有关物质检测结果的桥接研究。需要说明的是,虽然ICH Q3A[12]、ICH Q3B[13]不适用于临床试验期间, 但鼓励申请人参考ICH Q3A、ICHQ3B 等指导原则对超过鉴定限的杂质、稳定性期间出现的超过鉴定限的杂质进行归属鉴别,各杂质水平不得超过已有的动物安全性试验支持水平。杂质清除转化研究是支持杂质控制策略的一项重要研究内容,申请人可以根据临床试验进展开展此项研究。
3.3 致突变杂质
前期调研中,Pre- Ⅲ期药学沟通案例提出致突变杂质问题的比例约为7%。通常情况下,致突变杂质的研究需参照ICH M7[14]开展并制定控制策略。用于晚期肿瘤患者的药物可参照ICH M7和ICH S9[15] 进行致突变杂质的评估和控制。如果早期开发用于晚期肿瘤,随着临床研究的进展和开发计划,如拟开发适应症为非晚期肿瘤(如一线用药)或其他非肿瘤适应症,需重新评估拟定的致突变杂质控制策略是否还适用。
另外,药审中心于2020 年发布了《化学药物中亚硝胺类杂质研究技术指导原则(试行)》[16],美国食品药品监督管理局(FDA)于2021 年发布了亚硝胺杂质的控制指导原则[17],欧洲药品管理局(EMA) 分别于2020 年、2023 年发布了亚硝胺杂质的评估报告[18] 及问答文件[19]。申请人在开展亚硝胺杂质研究过程中,可以参考上述指导原则进行评估,制定合理的控制策略。
3.4 晶型
对于固体制剂、半固体制剂、混悬剂等剂型来说,晶型对制剂生产工艺、稳定性以及制剂的溶出行为、药代动力学和疗效均可能存在影响。
在早期产品开发时需开展晶型研究,包括晶型筛选、晶型溶解度与溶解速率、结晶工艺开发、晶型稳定性等。需说明选择目标晶型的依据,以及在原料药粉碎、制剂生产及贮藏过程中晶型的稳定情况。原料药经干燥、粉碎、制粒和压片等制剂工艺步骤,在温度和湿度等环境因素的作用下,均可能出现转晶现象,进而影响制剂的溶出行为和(或)生物利用度。因此,通常采用热力学稳定的晶型进行开发,注意避免采用混晶或生产、贮藏过程中发生转晶后形成混晶。
建议参照ICH Q11 等指导原则,加强原料药结晶工艺研究及生产工艺参数的控制,保持原料药批间晶型的一致性。
3.5 原料药的粒度
原料药的粒度是原料药的一项关键质量属性,特别是难溶性的药物,原料药粒度对制剂的混合均匀度、溶出行为及生物利用度均有重要影响。一方面,可通过加强原料药结晶、粉碎步骤工艺参数的控制,包括但不限于结晶溶剂的用量/ 比例、结晶温度、升降温速率、粉碎参数等,从而控制原料药粒度在拟定范围内。另一方面,粒度测定方法应专属、灵敏,必要时可考虑与显微镜检测结果进行对比。临床试验期间需持续积累粒度检测数据,结合粒度对制剂关键质量属性(如混合均匀度、溶出行为等)及体内暴露行为的影响,合理制定控制策略。
3.6 溶出度/ 释放度研究
溶出度/ 释放度是口服固体制剂的一项关键质量属性。拟定的溶出度/ 释放度方法需有适度的区分力。溶出度/ 释放度方法开发时,建议根据药物在37℃的pH- 溶解度曲线,选择具有适度区分力的溶出介质/ 释放介质,并对装置(桨法/ 篮法等)、介质体积、转速等进行筛选,如需加入表面活性剂,需论证表面活性剂加入的必要性和加入量的合理性,对其种类、来源、用量等进行筛选,并开展方法的区分力研究,可以包括对处方变量、工艺参数变量、原料药粒径、晶型等的区分力。Ⅲ期临床试验期间需注意积累数据。
3.7 批量
《技术要求》中,批量问题并未作为单独的共性问题进行讨论,但实际工作中批量问题经常出现在沟通交流问题清单中。通常情况下,普通口服固体制剂的关键临床批次样品的批量建议不低于10 万片/ 粒,如已确定为罕见病用药,这类品种的批量可以及时与审评机构进行讨论确定合理批量。其他剂型的批量可以参照《化学仿制药注册批生产规模的一般性要求(试行)》[20] 等指导原则来制定。
3.8 临床试验期间药学变更
临床试验期间发生的药学变更可以参照《创新药(化学药)临床试验期间药学变更技术指导原则(试行)》开展研究,并按照《药品注册管理办法》相关要求,选择合适的申报路径。
需要特别关注的是,皮肤外用制剂、复杂制剂(如微球/ 微乳、脂质体、混悬型注射液等),由于其处方工艺、制备工艺相对比较复杂,对生产设备的要求比较高,如果发生生产场地、处方工艺、批量的变更,仅通过药学对比研究无法证明变更前后产品质量一致。因此,建议这类复杂制剂的生产场地、处方工艺、批量等的变更尽量在关键临床试验前开展。上市申请的生产场地、处方工艺、批量需与Ⅲ期临床试验保持一致。
4、 结 语
随着药品审评审批制度改革的不断深入,药品研发注册的配套法规、指导原则及技术要求等不断完善,为药物研发注册提供了技术指导。药审中心坚持鼓励以临床价值为导向的新药好药、罕见病用药、重大传染病用药及公共卫生方面的临床急需药品的研发创新,加速完善药品审评审批标准体系建设。《技术要求》与《化学药品创新药Ⅰ期临床试验申请药学共性问题相关技术要求》《化学药品创新药上市申请前会议药学共性问题及相关技术要求》组成了化学药品创新药临床期间及上市前的沟通交流技术要求体系,服务药品创新。申请人可以通过运用这些技术要求,就重大技术问题及时提出沟通交流,加快药品研发上市。
引用本文
徐立华,周浩辉,赵一飞,王亚敏*.化学药品创新药Ⅲ期临床试验前会议的药学共性问题及审评考虑[J].中国食品药品监管.2023.08(235):32-37.