研发小故事
案件1:稀释还是吸附?
某一天的中午,在沂蒙山革命老区茫茫的大山深处,有个充满现代化的药品研发生产企业,在一台高效液相前,众人凝视着弯弯曲曲的图谱,像是在等待着命运最后的裁决。
“高,高了”,在计算机结果出来前,有着丰富经验的分析老师激动的喊着,这涉及数十万一次的放大生产,出现的每个异常事件都足以扣人心悬,含量终于合适了,众人悬着的心终于放了下来。
中间产品100.12%的含量只经历了常规的除菌过滤,到达灌针只剩下了88.37%,针对这一异常事件,制剂、分析及车间立即展开了调查。
制剂:
(1)项目小试开发,滤器吸附评估实验所使用的滤器与放大生产使用的一致,含量无吸附;
(2)项目小试开发,生产组件吸附评估实验所使用的生产组件与放大生产使用的一致,含量无吸附;
(3)项目小试开发,药液稳定性实验无异常,放大生产采用小试参数应不会对药液稳定性产生显著影响,且其它理化结果无异常;
结论:滤器、生产组件对产品无吸附作用,在生产控制参数下不会对药液的稳定性产生显著影响;
分析:
(1)含量测定采用的是进口注册标准,经多批参比制剂和自制制剂检测,从无出现过异常现象;
(2)本次放大生产系统适应性及对照无异常,且中间产品含量检测也无异常;
结论:本次使用的含量分析方法不会对药液的含量测定产生影响;
生产:
(1)重新核对原料药信息、称量及投料无异常,且中间产品含量结果已说明配制工序无异常;
结论:生产过程中的配制工序无异常;
问题分析:
通过从制剂、分析及生产过程中配制工序等全方位调查,最终锁定到了过滤、灌装工序。由于项目小试开发滤器吸附评估实验所使用的滤器与放大生产使用的一致,含量无吸附,放大生产与小试即使在过滤压力、过滤流速有所不同,但不至于会对含量产生如此显著的影响,通过排查分析,将目光锁定到了灌装工序。
我们研发人员联合质量保证部(现场QA和QA主管)及车间同事从无菌储罐到灌针进行了全方位排查,发现灌装罐(连接灌针的5L小罐)没有安装罐底阀,由于车间生产线是安装过滤器后在线灭菌的,这样的好处是保证产线的无菌保障水平,坏处是即使通过压缩空气的吹扫还是会有一定量的水残留在管道中,导致打药时药液从无菌储罐携带管道中的残留水直达灌针,致使灌针处含量显著下降至88.37%。
解决问题:
我们快速反应,检测灌装开始0.5L、1L、1.5L、2L、5L样品含量,含量依次为91.22%、98.64%、100.29%、99.85%、100.52%,通过含量结果分析,灌装开始1.5L含量已恢复正常,为保证产品符合质量标准,降低风险,设计弃药2L再进行罐装。
产线改造及验证:
(1)在灌装罐设计罐底阀门,药液到灌装量再开始灌装;
(2)验证生产线灭菌后的吹扫时间,将管道残留水降至可接受程度;
(3)通过灌装开始的含量检测,设计弃药量;
引言
细菌和细菌内毒素是对无菌药品质量影响最大的两个因素,若药品的无菌和内毒素无法保证会危及患者的生命安全,我国的《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》对保证无菌药品的无菌给出了详细的策略和方法,但是如何采用科学合理的生产工艺提高SAL(无菌保证水平)保证最终产品的PUSN(无菌保证值)达到≤10E-6 cfu,是值得我们每一个从业人员思考的问题。
本文就针对非最终灭菌产品所采用无菌生产工艺中的除菌过滤系统,通过产品的小试开发、放大生产及工艺验证进行系统性的考量,不足之处,还请业内同仁指出,共同学习。
1、除菌过滤系统概述
除菌过滤系统是指采用0.22微米(更小孔径或相同过滤效力,0.1微米的除菌级过滤器通常用于支原体的去除)的除菌级过滤器和输送药液的管道组成的无菌系统,常用于非最终灭菌的注射剂、无菌原料及滴眼剂的使用。
2、车间除菌过滤系统的设计
为降低产品除菌过滤的风险,车间除菌过滤系统常采用的有减菌过滤系统、冗余过滤系统及减菌加冗余过滤系统,极少有企业会采用单一滤器除菌过滤系统。
2.1单一滤器除菌过滤系统
整个过滤系统只有一个除菌级过滤器,过滤前的微生物污染水平应小于等于10cfu/100ml,过滤器应安装在接近无菌储罐的位置,无菌储罐与灌装点距离应较近。
优点:
(1)整个过滤系统只有一个除菌级过滤器,成本低且便于进行无菌装配;
缺点:
(1)对除菌过滤前微生物污染水平及中间产品抑菌性要求较高;
(2)对生产线的管道设计要求较高;
(3)无菌风险大,不符合现有除菌过滤技术及应用指南相关原则;
过滤器完整性:
除菌过滤器需通过过滤器完整性测试。
总结:
单一滤器除菌过滤系统无菌风险较大且不具备普适性,不建议采用。
2.2减菌过滤系统
减菌过滤系统是在非最终灭菌工艺生产的无菌制剂的除菌过滤工序前端采用孔径0.22(或以下)微米或0.45微米的过滤器,使产品除菌过滤前的微生物污染水平小于等于10cfu/100ml。
优点:
(1)除菌过滤工序前端采用孔径0.22(或以下)微米或0.45微米的过滤器,使产品除菌过滤前的微生物污染水平小于等于10cfu/100ml,降低了工艺对原辅料的微生物负荷及中间产品抑菌性的要求;
过滤器完整性:
减菌过滤器和除菌过滤器都需通过过滤器完整性测试。
总结:
减菌过滤系统是为了保证产品除菌过滤前的微生物污染水平符合可接受标准,还需关注产品的细菌内毒素控制策略,做到两者兼顾,最大程度的降低产品的无菌及细菌内毒素风险。
过滤前可见异物较多的产品,减菌过滤建议采用0.45微米的过滤器,防止阻塞过滤器;
过滤前可见异物较少的产品,减菌过滤建议采用0.22(或以下)微米的过滤器;
2.3冗余过滤系统
冗余过滤系统是指在最终除菌过滤器前增加一个除菌级过滤器。增加的这个除菌级过滤器即为冗余过滤器,冗余过滤系统的首级滤器应尽可能安置在无菌区域外且接近无菌储罐,而第二级过滤器(主过滤器)应接近灌装点。
除菌过滤前的微生物污染水平应小于等于10cfu/100ml,且两个过滤器之间必须确保无菌。
优点:
(1)整个过滤系统有两级除菌级过滤器,通过提高无菌生产工艺的无菌保证水平提高了最终产品的无菌保证值,使产品的无菌保证值远低于10-6 cfu;
缺点:
(1)对除菌过滤前微生物污染水平及中间产品抑菌性要求较高;
(2)过滤前产品的可见异物较多时,容易造成首级过滤器不同程度的堵塞,需控制原辅料的可见异物;
过滤器完整性:
主过滤器完整性测试通过,则冗余过滤器不需要进行完整性测试;如果主过滤器完整性测试失败,冗余过滤器必须通过完整性测试。
总结:
冗余过滤系统通过严控除菌过滤前微生物污染水平及中间产品的抑菌性,保证过滤前的微生物污染水平小于等于10cfu/100ml,兼顾了产品的细菌内毒素控制,提高了最终产品的无菌保证值。
2.4减菌加冗余过滤系统
减菌加冗余过滤系统是指在冗余过滤系统前端增加孔径0.22(或以下)微米或0.45微米的过滤器,使产品除菌过滤前的微生物污染水平小于等于10cfu/100ml,降低了工艺对除菌过滤前微生物污染水平及中间产品抑菌性的要求;
优点:
(1)整个过滤系统有一个减菌过滤器和两个除菌级过滤器,在降低工艺对除菌过滤前微生物污染水平及中间产品抑菌性要求的同时,通过提高无菌生产工艺的无菌保证水平提高了最终产品的无菌保证值,使产品的无菌保证值远低于10-6 cfu;
缺点:
(1)整个过滤系统共使用了三个过滤器,过滤成本较高;
过滤器完整性:
减菌过滤器需通过过滤器完整性测试。
主过滤器完整性测试通过,则冗余过滤器不需要进行完整性测试;如果主过滤器完整性测试失败,冗余过滤器必须通过完整性测试。
总结:
减菌加冗余过滤系统虽然可以显著的提高最终产品的无菌保证值,但无法截留细菌内毒素,在设计药液存放时限时需兼顾产品的细菌内毒素控制。
2.5过滤系统的选择
单一滤器除菌过滤系统对过滤系统的设计要求较高、无菌风险大,不符合现有除菌过滤技术及应用指南的原则,不建议采用;
根据对除菌过滤前微生物污染水平及中间产品抑菌性的理解,科学合理的选择过滤系统。
首选无菌工艺模拟试验验证过的过滤系统(法规要求);
(1)当中间产品过滤前微生物负荷小于等于10cfu/100ml且中间产品有一定的抑菌性,选择冗余过滤系统;
(2)当中间产品过滤前微生物负荷大于10cfu/100ml,中间产品无抑菌性,选择减菌过滤系统或减菌加冗余过滤系统;
3、小试研发除菌过滤系统验证
通过考察产品计划转入车间相应生产线的除菌过滤系统来设计小试研发除菌过滤系统的相关验证。
3.1验证项目:
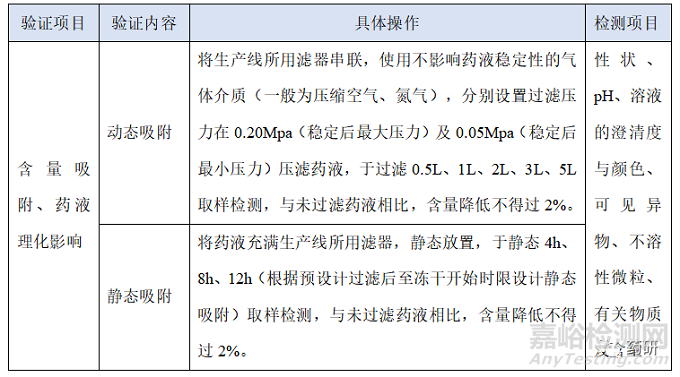
(1)除菌过滤系统对药液含量的吸附:包括动态吸附和静态吸附;
(2)除菌过滤系统对药液理化性质的影响:包括性状、pH、溶液的澄清度与颜色、可见异物、不溶性微粒及有关物质;
(3)除菌过滤系统对药液微生物的影响:除菌过滤前微生物污染水平、除菌过滤后无菌(小试一般不验证,由滤器厂家做细菌截留验证;中试、工艺验证做过滤前微生物污染水平,过滤后药液及成品的无菌检测);
3.2验证设计:
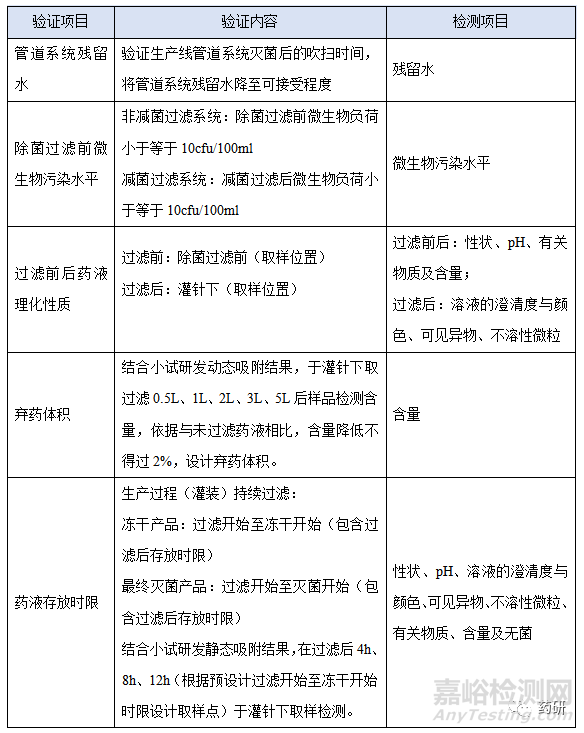
检测标准:
(1)含量: 动态吸附及静态吸附,与未过滤药液含量相比,不得过2%;
(2)其他项目:符合质量标准,不同取样点应无显著差异(过滤前后药液溶液的澄清度与颜色、可见异物、不溶性微粒除外);
4、中试除菌过滤系统验证
保证中试生产与小试验证参数一致性,尽量保证中试放大与工艺验证所用生产线一致性,这样中试除菌过滤系统验证既有小试参数做依据又为工艺验证除菌过滤系统验证提供参考。
4.1验证项目:
(1)除菌过滤系统对药液含量的吸附:动态吸附;
(2)除菌过滤系统对药液理化性质的影响:包括性状、pH、溶液的澄清度与颜色、可见异物、不溶性微粒及有关物质;
(3)除菌过滤系统对药液微生物的影响:微生物污染水平、无菌(过滤前微生物污染水平,过滤后药液及成品的无菌检测);
4.2验证设计:
检测标准:
(1)管道系统残留水:降至可接受程度;
(2)除菌过滤前微生物污染水平:小于等于10cfu/100ml;
(3)过滤前后药液理化性质:与未过滤药液相比,含量降低不得过2%;其他项目:符合质量标准,过滤前后应无显著差异(过滤前后药液溶液的澄清度与颜色、可见异物、不溶性微粒除外);
(4)弃药体积:与未过滤药液相比,弃药后正常灌装产品含量降低不得过2%;
(5)药液存放时限:各检测项目应符合质量标准,不同取样点应无显著差异;
5、除菌过滤验证
中试放大除菌过滤系统验证结束后若无异常,需委托滤器厂家进行除菌过滤工艺验证。
除菌过滤工艺验证一般包括化学兼容性试验、可提取物或浸出物试验、细菌截留试验(含细菌生存性试验)、产品润湿的完整性测试,吸附评估(一般由制剂公司做)及安全性评估等内容,应结合产品特点及实际过滤工艺中的最差条件,对相关验证进行科学合理的设计。
6、工艺验证除菌过滤系统验证
除菌过滤验证通过后,依据中试放大除菌过滤系统验证的相关参数进行工艺验证除菌过滤系统验证,验证项目及验证设计同中试除菌过滤系统验证一致。
7、参考资料
1、化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)
2、《中国药典》( 2020年版)中国医药科技出版社, 2020.
3、除菌过滤技术及应用指南
4、无菌工艺模拟试验指南(无菌制剂)
5、EU.GMP Annex 1 : Manufacture of Sterile Products(2020).
6、FDA Guidance for Industry Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice.
7、PDA technical Report No. 44 Quality risk management for aseptic processes(2008).
8、PDA Sterilizing Filtration of Liquids Report No. 26 (2008 Revision) of PDA.