摘要
基因毒性杂质是受到药品监管机构和制药企业重点关注和控制的对象,由于其较一般杂质具有微量水平就存在潜在致突变性和致癌性风险的特点,需要严格控制其在药物中的含量以保证药物质量与临床应用的安全性。本文从识别警示结构、文献及数据库检索、量化构效关系软件预测、体内外遗传毒性试验、依照法规和指南要求制定合理限度等方面综述了评估杂质基因毒性的研究策略,并总结了目前为了应对其含量低,稳定性较差,结构多样等特点开发的各类前处理技术及分析方法,为基因毒性杂质的研究提供参考。
背景
基因毒性杂质( genotoxic impurity,GTI) 是指药物中能直接或间接导致 DNA 受损引起基因突变,并具有致癌性或者潜在致癌可能性的一类杂质[1]。为了保证药物的质量安全,2002 年欧洲药物管理局( EMA) 最先出台了关于基因毒性杂质的管理法规,美国食品药物管理局( FDA) 、国际人用药品注册技术要求国际协调会( ICH) 等组织也针对基因毒性杂质先后颁发了相关界定、分类、限度、检测和风险评估程序等一系列指南,目前已经成为药品获批及上市的关键指标之一。欧洲药品质量管理局( European Directorate for the Quality of Medicines & Health Care,EDQM) 每年发布的上一年度申报资料中存在的十大常见缺陷问题报告中曾多次提到企业缺乏对基因毒性杂质的讨论。提出的问题包括: ①缺少对工艺中使用到的起始物质、溶剂、中间体、试剂等可能引入基因毒性杂质的讨论; ②缺少对终产物中基因毒性杂质的危害性评估; ③缺少对产品中基因毒性质控制的讨论。为解决以上问题,本文通过概括基因毒性杂质的研究思路,从其分类、危害评估方法、限度及分析方法建立等方面进行分析和总结,为基因毒性杂质的控制提供一定的理论依据和参考。
药物中基因毒性杂质的具体研究思路如下:①通过对药物合成、生产及贮存过程进行分析,结合警示结构,鉴别并分离出其中可能引入的基因毒性杂质; ② 通过数据库文献检索、( 定 量) 构 效 关 系[( quantitative ) structure- activity relationships,QSAR]评估和体内外相关毒理学试验对可能存在的基因毒性杂质进行分类; ③结合相关指南及法规,确定基因毒性杂质可接受的限度标准; ④根据基因杂质的理化性质及结构特点,建立理想的分析方法,并进行方法学验证; ⑤对药品进行检测,确定其中基因毒性杂质含量是否符合要求。
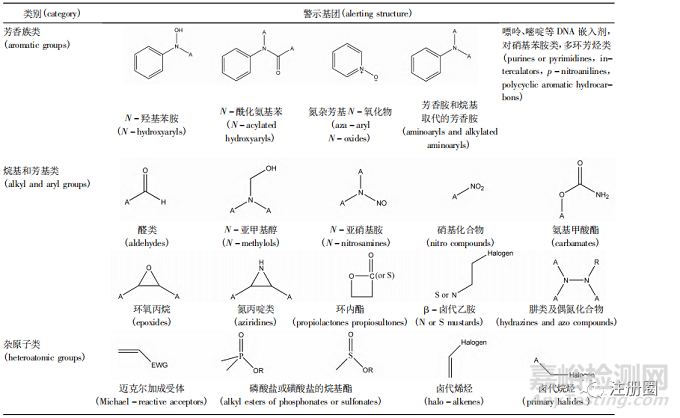
一、基因毒性警示结构
基因毒性化合物多种多样,其结构中通常含有某些可能诱导基因突变或癌症发生的特殊基团,被称为警示结构。在缺乏安全性数据支持的情况下,目前的法规和指导原则均采用警示结构作为普通杂质和基因毒性杂质的区分标志。目前美国国立卫生研究院数据库已经发布了 1 574 种致癌物质的结构式、CAS 号等常用信息,常见的警示结构主要有 3类,见表1[2]。然而,警示结构的意义仅在于它对于可能存在遗传毒性和致癌性的杂质起到了基础的提示作用,需要结合杂质的理化性质及其结构特点,为其危害评估与进一步的限度控制提供方向。
▲ 表1-基因毒性杂质警示结构
Tab. 1 Alerting structures of genotoxic impurities
注(note) : A. 烷基、芳基或氢原子( Alkyl,Aryl or H) ; Halogen. 卤素原子( F,Cl,Br,I) ; EWG. 吸电子基团( electron withdrawing group)
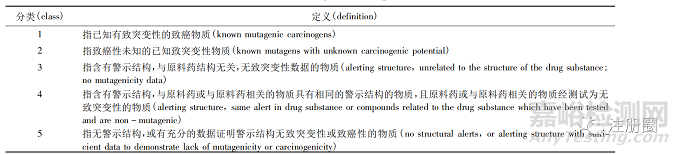
二、遗传毒性杂质的危害评估方法
目前根据致突变和致癌风险危害程度可将杂质分为5 类,见表2[3]。对于1 个未知的、可能具有潜在基因毒性的化合物,其危害评估应通过数据库文献检索、QSAR 评估以及体内外遗传毒性试验等评估方法对化合物进行分类,再根据分类制定具体的可接受限度标准。
▲ 表2-基因毒性杂质分类
Tab. 2 Classification of genotoxic impurities
2. 1 数据库、文献检索评估方法
在鉴定1个杂质是否为基因毒性杂质时,首先可以通过查询相关数据库中毒理学事实数据快速有效地对杂质进行分类,从而避免不必要的动物试验,节省成本。可用于判断化合物基因毒性的数据库包括: ①毒理学数据库(TOXNET) ( https: / /toxnet. nlm. nih. gov /) ,该数据库由美国国立医学图书馆建立,其中 TOXLINE为文献型数据库,危险化合物数据库(HSDB) 、遗传毒理学(GENE-TOX) 、化学物质致癌作用研究信息系统(CCRIS) 等为事实与数值型数据库,均可用于检测化合物的致突变性以及致癌性[4]。②The carcinogenic potency database ( https: / /toxnet.nlm.nih.gov /new toxnet /cpdb.htm) 为 TOXNET的子数据库,收录了1980-2011年间发表过的文献或由美国癌症研究所和国家毒理学计划报告过的致癌性化合物相关数据,共收录 1547 种致癌性化合物[5]。③化学物质毒性数据库( https: / /www.drugfuture.com/toxic /) ,该数据库收录了约15 万个化合物的毒理方面的数据,包括遗传毒性、致癌以及生殖毒性并提供数据来源,可供多种方式查询,包括化学名称、CAS 登记号、英文名等。
除了以上几个可专门查询化合物相关毒性的数据库,其他数据库如SciFinder 或 PubMed 等也可以查询相关毒理学数据。如果目前未收录待测化合物体内外相关实验数据,可结合 QSAR 对其基因毒性进行进一步的评估。
2. 2 QSAR 评估方法
QSAR评估方法是根据化合物结构特性与致突变性和致癌性的定量相关性来对待测化合物的基因毒性进行预测。EMEA 和 FDA 指南草案均认为QSAR是一种评估潜在基因毒性的有效方法。应用QSAR方法进行评估时,应采用2个预测原理互补的QSAR预测方法: 1个方法基于专家规则,另1个方法基于统计学,专家规则中包含的知识基于统计学大量数据证明,而统计学规则中的分子描述符、模型的建立和数据的收集则应由专家指导[3]。
2. 2. 1 基于专家规则
基于专家规则的评估软件是通过收集现有数据(已发表的文献、公开或内部数据库等) 建立解决问题所需的知识库,从而模拟具有化学和遗传毒理学专业知识和经验的专家的判断和行为。专家规则软件的 1个特点是能够解释自己的推理并提供相关证据,交由使用者来理解和判断所得出的结果的合理性。如有必要,所有基于计算机系统的分析结果均可以使用专家知识进行回溯,为所有预测的阳性、阴性、相互矛盾或无法得出结论的结果提供额外的支持性证据,从而保障最终结论的合理性[3]。目前使用较为广泛的基于专家规则的软件有DEREK、Leadscope Expert Alerts、MultiCASE、ToxTree 等。
2. 2. 2 基于统计学
与专家规则不同,基于统计学的软件是通过收集细菌致突变性试验数据及相关毒理学文献,建立起一类基于化合物结构和基因毒性之间相关性的统计学模型。输入待测物的结构式,软件可以通过计算参数、结构相关性以及各类统计方法来预测其潜在基因毒性。数据库除了可以预测化合物整体结构的毒性,还可以给出化合物结构中某个官能团对毒性的贡献水平。目前常用的基于统计学规则的软件有Leadscope、Sarah Nexus、TOPKAT、Multi-CASE、Case Ultra 等。
获得 2 种 QSAR 方法的评估结果后需要根据其给出的证据进行分析。当 2 种方法判断该杂质均为阳性时,表明该杂质有很大几率为 1 类或 2 类杂质,除非有更为权威的数据证明其为 4 类杂质; 如果 2 种方法的结果均为阴性,表明该杂质有可能为 4 类或 5类杂质,或数据不足以判断为基因毒性杂质; 如果 2种方法的结果 1 个为阳性而另 1 个为阴性,或者出现模棱两可及超出判断范围的结果,则可以将其评估归为 3 类杂质,并进一步开展遗传毒性试验。祝清芬等[6]采用 Derek( 专家知识规则) 和 Sarah( 统计学)2 类 QSAR 评价软件,对左羟丙哌嗪中的 3 个杂质以及羟苯磺酸钙中的 1 个杂质进行基因毒性评价和分类。其中缩水甘油被 Derek 判断具有环氧化物警示结构,结合 Sarah 列举相关文献和数据库匹配的支持性结构判断其遗传毒性为阳性的可信度为 100% ; 另1种杂质氢醌虽然在 Derek 中预测结果为阴性,但是结合 Sarah 提供的阳性支持性结构和文献判断其遗传毒性定为阳性。已知缩水甘油和氢醌分别被国际癌症研究所( IARC) 列为 2A 类和 3A 类致癌物,说明基于指导原则的要求使用 QSAR 软件可以对杂质做出合理的遗传毒性预测。
2. 3 遗传毒性试验
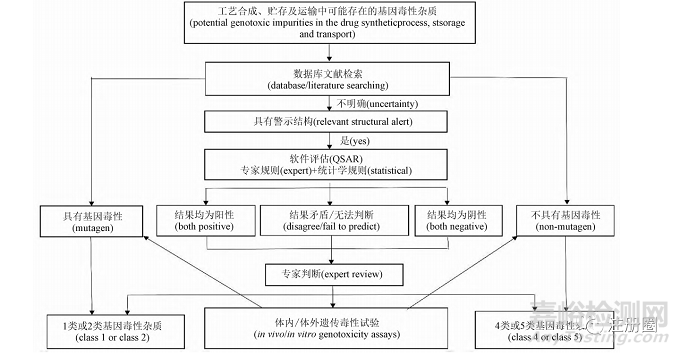
遗传毒性试验是指用于检测受试物通过不同原理直接或间接导致遗传物质损伤的体内外试验,遗传物质的损伤包括基因突变、染色体数目的改变、染色体损伤或重组导致的 DNA 损伤等,对于基因毒性不明确的杂质,应进一步进行遗传毒性试验评估。如果遗传毒性试验结果呈阳性,在结合其他药学研究数据排除了假阳性的情况下可判断该杂质具有遗传毒性,为 1 类或 2 类杂质,若为阴性,则可判断该杂质为 4 类或 5 类杂质,见图 1。试验方法可分为体内和体外 2 种类型,体外试验包括细菌回复突变试验( Ames 试验)、体外中期相染色体畸变试验、体外微核试验、小鼠淋巴瘤细胞试验( mouse lymphoma assay,MLA) 等,可选择的试验材料包括从原核细胞、真核细胞和高等哺乳动物细胞等; 体内试验包括体内骨髓微核或外周血红细胞微核试验、骨髓细胞染色体变试验、单细胞凝胶电泳试验( 彗星试验) 、转基因小鼠体内突变试验、外周淋巴细胞测定染色体畸变试验、DNA 共价结合试验等,试验材料多采用啮齿类动物[7]。以上实验均已得到验证并被广泛应用。
▲ 图1-基因毒性杂质危害性评估步骤
Fig. 1 Hazard assessment steps of genotoxic impurities
遗传毒性试验的设计应根据受试物特点选择合理的方案,为制定合理杂质限度标准提供可靠数据。目前,为了全面评估待测物的遗传毒性风险,通常采用体内和体外试验相互补充的方法对待测物的遗传学终点进行全面考察,其中体外试验应包含 Ames 试验并且至少采用 5 种菌株。除此之外,由于体内试验具有更贴合临床实际使用情况的优势且可检出体外试验无法检出的某些遗传毒性化合物,因此标准试验组合中应至少包含 1 项体内试验,在药物遗传毒性研究指导原则中推荐了 2 种标准试验组合,见表 3。推荐组合并不代表其更优,可以根据待测物具体生物活性特点合理采用其他经过验证的试验作为替代试验,用作对标准试验组合得到的遗传毒性试验结果的进一步研究。
▲ 表3-推荐标准实验组合 Tab. 3 Standard combinatorial tests
注(note) : + . 需做( request the test) ; ± . 视情况而定( depend on the situation)
在完成一组体内外标准试验组合后对结果进行分析,若试验结果为阴性,通常可提示受试物缺乏遗传毒性。对于得到阳性结果的待测物,应结合其他药学研究数据进行综合分析。Asuka等[8]对谷氨酸钠进行了一系列遗传毒性试验以研究其致突变性和致癌性,包括 Ames 试验、体外染色体畸变试验、小鼠淋巴瘤tk试验、人外周血淋巴细胞体外微核试验以及和大鼠骨髓微核试验。与对照组相比,谷氨酸钠对任何细菌株均无致突变性,无致染色体畸变、基因突变或致微核作用,判断谷氨酸钠在研究条件下不具有遗传毒性。
三、限度计算
为了控制药品中基因毒性杂质的含量,EMA 2006 年颁布了《基因毒性杂质限度指南》,引用毒理学关注阈值 ( threshold of toxicological concern-based,TTC) 这一概念界定所有未经研究,但具有致癌风险或其他毒性效果的化合物的可接受摄入量,基因毒性杂质的可接受限度为1. 5 μg·d-1[9]。TTC作为风险评估指标,不能理解为绝对无风险,部分化合物已被证实具有较高的致癌风险,因此需要制定较 TTC 更为严格的控制标准。各个国家及制药企业的研究学者提出了“阶段化TTC”的概念,即原料药中的基因毒性杂质限度应当考虑药物的暴露周期,暴露周期较短的药物应当可以设定较高的控制限度。2017年5月31日 ICH出台了第4版指南 M7( R1) ,在阶段性 TTC 策略的基础上进一步调整和增补相关指导原则,使得研究人员能够根据药物暴露周期,科学地控制基因毒性杂质含量,使风险评估更 加灵活准性[3]。在实际制定某一 GTIs 的可接受标准时,应根据 GTIs 致突变和致癌风险危害类别以及其现有毒理学数据选择合适的计算方法,杂质限值的计算公式一般为杂质限值 = 杂质每日可接受摄入量 /药物每 日 最 大 用 量。其中可接受摄入量计算方法见表 4,对于无毒理学研究数据的杂质可采用 TTC 计算可接受摄入量,其计算方法分为单个杂质和多个杂质,见表 5。
以N-二甲基亚硝胺( NDMA) 为例,NDMA 为第1类杂质,在小鼠与大鼠中的TD50值分别为 0. 189 mg·kg-1·d -1和 0. 095 9 mg·kg-1·d-1( 按照更为保守的大鼠TD50值0. 0959 mg·kg -1·d-1计算) ,人体质量50 kg,风险概率按肿瘤发生风险为十万分之一来计算每日摄入量可得[3]: 每日摄入量( PDE) = TD50 × W人体质量 /50 000 =0. 095 9 ×50/50 000 =0. 095 9 μg·d-1。
对于缺少致癌性数据,但化学结构与已知致癌物类似的致突变杂质,可以采用化合物特异性的方法来计算致突变杂质的可接受摄入量,ICH M7( R1)中详细列举了不同分类下 GTIs 的每日可接受摄入量计算方法。在明确了GTIs的限度后,可以根据要求 开发适宜的分析方法,并对其进行优化及验证。
▲ 4-基因毒性杂质可接受摄入量的计算方法
Tab. 4 Counting method of acceptable intakes of genotoxic impurities

四、分析方法的建立及策略
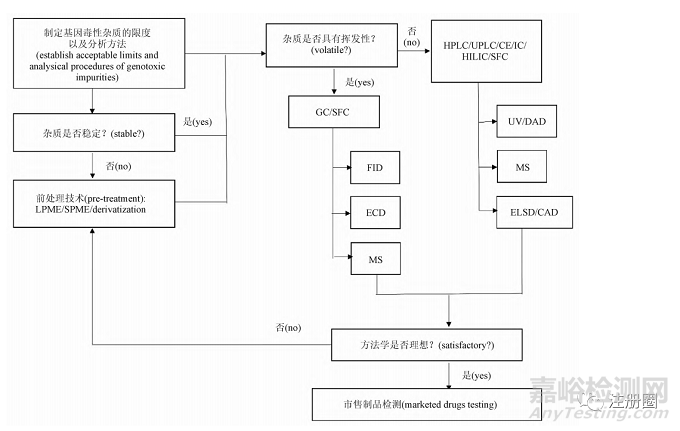
与一般杂质相比,基因毒性杂质特点是在微量水平即可造成人体内遗传物质的损伤,因此 GTIs 的分析检测是一项具有挑战性的研究。目前药物中基因毒性杂质限度通常被控制在非常低的水平,对分析仪器的灵敏度要求较高; 部分 GTIs 化学反应活性较高或稳定性较差,在提取、制备或者分析过程中易发生反应或挥发,导致回收率低,准确度差; 除了灵敏度和准确度之外,分析方法还应具有能将 GTIs 与一般杂质分离开的高选择性。为了应对这些挑战,需根据不同基因毒性杂质的特点探索各种分析策略,选择合理的样品前处理手段和分析仪器,开发并优化分析方法,以便准确地测量和控制药品中 GTIs水平,分析思路见图 2。
4. 1 样品前处理技术
恰当的前处理手段可以有效减少样品损耗,提高分析方法的选择性以及灵敏度,以便获得更好的检测结果。这些前处理手段可以使样品的纯化、富集和检测一体化,优化检测效率。
4. 1. 1 萃取技术
由于基因毒性杂质检测很低,检测时有可能引入原料药或者溶剂杂质,导致无法得到准确的结果,需要采用适当的萃取技术,对样品进行分离纯化和富集,从而满足进样标准,在减少基质的干扰及提高检测灵敏度的同时可以与气相色谱或液相色谱在线联用,具有萃取效率高及操作简单等优点。
4. 1. 1. 1 夜-液萃取法及液相微萃取法
液液萃取( LLE) 是最为传统的一种萃取方式,欧洲药典( EP) 采用 LLE - 顶空进样(headspace,HS) - GC/MS 法测定药物中的硫酸甲酯( MMS) 、硫酸乙酯( EMS) 和硫酸异丙酯( IMS)[10]。但是 LLE法操作烦琐,萃取溶剂毒性大且用量较多,容易发生乳化,从而影响测定结果。1996 年液相微萃取( LPME) 技术出现,具有萃取效率高及溶剂消耗少的优点,可以与色谱仪在线联用,实现样品萃取、富集和检测一体化。Cui 等[11]采用了分散液相微萃取作为前处理手段去除基质效应,并联用HPLC-UV 法测定酒石酸依格列司他原料药中的1,4-苯并二 烷-6-甲醛,检测限为1. 29 μg·g-1。Durak 等[12]采用可切换溶剂的液相微萃取技术作为样品前处理和富集手段,联用 GC - MS 法对4个氯化有机物进行检测,其检测限较直接采用GC-MS分析样品的方法提高了33~115倍。
▲ 图2-基因毒性杂质分析方法建立思路
Fig.2 Analytical procedures of genotoxic impurity
HPLC. 高效液相色谱( high performance liquid chromatography) UPLC. 超高效液相谱(ultraperformance liquid chromatography) CE. 毛细管电泳色谱( capillary electrophoresis) IC. 离子色谱( ion chromatography) HILIC. 亲水作用色谱( hydrophilic interaction liquid chromatography) SFC. 超临界色谱( super critical fluid chromatography) GC. 气相色谱( gas chromatography) FID. 氢火焰离子化检测器( flameionization detector) ECD. 电子捕获检测器( electron capture detector) MS. 质谱( mass spectrometry) UV. 紫外吸收检测器( ultraviolet absorption detector) DAD. 二极管阵列检测器( diode array detector) ELSD. 蒸发光散射检测器( evaporative light scattering detector) CAD. 电喷雾检测器( corona charged aerosol detector) LPME. 液相微萃取技术( liquid phase microextraction) SPME. 固相微萃取技术( solid phase microextraction)
4. 1. 1. 2 固相萃取法及固相微萃取法
固相萃取(SPE) 是一种利用固定相对分析组分的吸附作用,将待测物从基质中萃取并富集的前处理过程,一般选择具有良好吸附性能的材料如碳纳米管、分子印迹聚合物等颗粒固相进行萃取,待测物检测限也随着固相成分的发展大大降低,例如以吩噻嗪键合硅胶为固相联合GC-MS进行分析[13],硝基苯类化合物的检出值可以低至0. 06~0. 3 ng·mL-1。Tzanavaras等[14]通过在线固相萃取技术分离并测定别嘌呤醇中肼的含量,以邻苯二甲醛作为衍生化试剂在酸性条件下与肼反应形成腙衍生物,利用亲水吸附剂成功地将待测物从别嘌呤醇中分离,避免了美国药典中提出的溶剂萃取过程并提高了灵敏度。与固相萃取技术相比,固相微萃取( SPME) 技术更加高效简便,可以采用纤维针式 SPME、管内 SPME、固态搅拌棒萃取等多种形式从复杂基质中富集待测物进行检测,在药物分析领域中应用广泛。Ivelisse等[15]采用固相微萃取-气相色谱/质谱联用技术,在选择离子监测模式下完成了原料药中 7个磺酸酯类基因毒性杂质的测 定,并 比较了Carboxen /聚二甲基硅氧烷(CAR/PDMS) 、聚二甲基硅氧烷/二乙烯基苯( PDMS /DVB) 以及聚乙二醇/二乙烯基苯(CW/DVB) 等固相微萃取萃取头之间的差异。由于PDMS /DVB能更有效地从原料药中提取磺酸酯类基因毒性杂质,与质谱联用进样时灵敏度更高,重现性更好,因此作者选择其作为萃取头材质,并对微萃取时间以及提取介质的 pH 等条件进行了优化。最终该方法可以有效检测原料药中5 ng·g-1左右的磺酸酯类基因毒性杂质,所得萃取溶液中磺酸酯类杂质的浓度为50 ng·g - 1左右,方法重现性较好。
4. 1. 2 衍生化技术
衍生化技术是一种利用化学反应改变难以分析检测的待测物的化学和物理性质,定量地转化成另一种更易分离检测的化合物的前处理技术。通过衍生化可以改善杂质的色谱特性和分离效果,提高方法的选择性和灵敏度,同时对检测过程中不稳定的待测物起到保护作用。应根据不同类型GTIs 的理化性质或限度要求选择合适的衍生化试剂进行反应。
4. 1. 2. 1 磺酸酯类化合物
由于酯类易水解的特点,磺酸酯类化合物通常采用衍生化的手段将其转化为较为稳定的化合物,以有效提高检测结果的准确性。EP提供了一种以碘化钠作为衍生化试剂,采用顶空进样GC/MS法检测甲磺酸倍他司汀、苯磺酸氨氯地平和对甲苯磺酸舒他西林原料药中的痕量甲磺酸酯、苯磺酸酯和对甲苯磺酸酯的方法,因操作简便而受到广泛的应用。Zhou 等[16]使用衍生化试剂N,N-二乙基二硫代氨基甲酸酯(DDTC) 向待测甲磺酸甲酯和甲磺酸乙酯中引入生色基团,使其适用于HPLC- UV作为分析方法。Li 等[17]通过碘化钠将甲磺酸伊马替尼和甲磺酸左氧氟沙星原料药中的GTIs 甲基磺酸甲酯、甲基磺酸乙酯和甲基磺酸异丙酯衍生化为相应的碘化烷烃,经顶空单液滴微萃取法富集后再使用 DDTC 衍生化,经 HPLC- UV分离检测,检测限最低为15 ng·mL-1。
4. 1. 2. 2 肼类化合物
肼类化合物具有活泼性高,挥发性强,分子量小,不具备发色基团等特殊化学性质,且容易与色谱柱中的硅氧基生反应从而导致峰形不理想,因此常采用衍生化法手段减少药物基质的干扰,提高稳定性和结果准确性。Wang等[18]采用2-羟基-1-萘醛作为衍生化试剂与肼反应生成腙类化合物,减少基质干扰的同时提高了方法选择性和灵敏度。卡巴克洛( CBZ) 和卡巴克洛磺酸钠( CSS) 中都含有氨基脲基团,合成过程中可能会引入肼类基因毒性杂质,Canavesi 等[19]开发了一种LC-DAD 法用来测定CBZ 和 CSS 药物中氨基脲盐酸盐含量的方法,采用 2 - 萘甲醛作为柱前衍生化试剂将氨基脲转化为具有发色基团的 2-萘甲醛氨基脲( NCS) ,并联 LLE 富集纯化,检测限可达到0. 02 μg·mL-1。
4. 1. 2. 3 卤代烷烃类及酰氯类化合物
卤代烷烃种类繁多且性质各不相同,对于非挥发的烷基卤化物,可通过衍生化法改善其挥发性,进而采用GC法进行测定。例如氯乙醇和1- 氯丁醇沸点较高,不适合气相色谱法检测,结构中只有1个氯原子取代也使 ECD的灵敏度和回收率较低。David 等[20]采用七氟叔丁基氯对氯乙醇和1-氯丁醇进行衍生,引入了多个氟原子,使样品杂质的挥发性增加,提高其在ECD 中的检测灵敏度,其检测限为5 ng·mL-1。Bai等[21]开发了一种以二甲胺作为衍生化试剂下与 2 种氯代烷烃反应引入胺基的衍生方法,有效提高了其灵敏度,1 ng·mL-1 浓度下其信噪比为 30 和 36。Zheng等[22]建立了一种测定药物中芳香族化合物和脂肪族酰氯的衍生化 HPLC-UV 法,并且对比了硝基苯胺和硝基苯肼2种衍生化试剂; 结果表明,硝基取代苯肼衍生物显示出最大波长和吸光强度的增加,可以有效减少基质干扰,提高了方法的特异性和灵敏度,检测限范围在0. 01~ 0. 03 μg·ml-1。
4. 1. 2. 4 环氧类化合物
环氧化合物中三元环通常不稳定,常在气相色谱样品入口处受高温影响而开环降解,导致分析过程精密度和准确度较差。衍生化是解决以上问题常用的方法。Bai 等[21]报道了一种利用二甲胺作为衍生化试剂使环氧化物形成稳定结构的方法,并利用衍生化物的高质子亲和力采用液相色谱 - 电喷雾质谱法(LC-ESI-MS) 进行检测,提高了灵敏度,2种环氧化物信噪比在5 ng·mL-1和7. 5 ng·mL-1浓度下分别为 50 和 260。Wu等[23]报道了一种利用 Meerwein 反应进行衍生化,再联合LC- MS 测定环氧化合物含量的方法,检测限为10 ng·mL-1; 研究发现以乙基氮杂炔正离子作为衍生化试剂与环氧化合物反应,可以增加环氧化合物的稳定性,酰基正离子也显示可以与环氧类化合物发生相同的反应。
4. 1. 2. 5 芳香胺类化合物
芳香胺类化合物分子结构中与苯环相连的氮原子经代谢活化,易生成稳定的氮正离子,能与 DNA 结构中的亲核活性基团发生反应,因此具有潜在致突变性。除此之外芳香硝基类化合物缺乏足够的电离效率,可通过化学衍生化将其转化为带电荷的芳香胺类化合物,再进行液相色谱或气相色谱联用质谱法进行痕量分析检测。Wu 等[24]选择利用 50% 甲酸铵溶液作为2- ( 溴甲基) - 6-硝基苯甲酸甲酯的衍生化试剂,锌粉作为催化剂,摇匀后离心取上清液进行LC-MS 分析,检测限低至 0. 2 ng · mL - 1,回收率均 在80% 以上。Vanhoenacker 等[25]开发了一种检测原料药中13 类芳香胺和氨基吡啶的原位衍生化 LC-MS方法,并对邻苯二甲醛、9-氟甲氧羰基氯、氯甲酸己酯、3,5 -二硝基苯甲酰氯等几种常用胺类衍生化试剂进行研究,结果表明氯甲酸己酯作为衍生化试剂可获得较好的灵敏度和回收率。
化学衍生化法目前广泛用于各类药品相关物质分析研究中,除了其带来的优点之外,也有不可忽视的缺点,如操作过程烦琐,重现性差,衍生化试剂形成的副产物可能对色谱分离造成较大困难,引入杂质或干扰峰,造成样品损失等。因此,选择适当的前处理技术是建立理想的分析方法的前提,为下一步选择合理的分析检测仪器打下基础。
4. 2 高效液相色谱法
高效液相色谱法( HPLC) 是检测不易挥发或热不稳定 GTIs 的常用方法,通常需要从样品处理、色谱柱、柱温、流动相、梯度比例、检测器等各方面对方法进行优化,以满足 GTIs 的痕量检测需求。其常联用的检测器包括紫外检测器、二极管阵列检测器、质谱检测器、带电气溶胶检测器和蒸发光散射检测器等。
4. 2. 1 紫外检测器和二极管阵列检测器
紫外检测器( UV 检测器) 作为最常见的液相色谱联用检测器,具有灵敏度高,选择性好和可检测范围广的特点,由于其简单且有效,对于非挥发性 GTIs一般可将高效液相色谱 - 紫外检测法( HPLC - UV)法作为首选。二极管阵列检测器( DAD) 原理与 UV检测器相同,并且能够进行全波长扫描、检测峰纯度等操作,在建立不确定检测波长的 GTIs 分析方法时具有优势。Griˇcar 等[26]建立了一种反相 HPLC - UV法以梯度洗脱测定沙坦类药物中叠氮化物含量,在波长 205 nm 处进行紫外检测,其检测限可以达到0. 84 μg·g - 1,具有良好的选择性和准确性。芳基磺酸酯类 GTIs 沸点较高,不宜用 GC 法测定,通常采用HPLC - DAD 法 测 定。García 等[27] 开 发 了 反 相HPLC - DAD法用来检测氯哌斯汀芬地柞酸原料药中的对甲基苯磺酸甲酯和 2 - 氯乙基对甲苯磺酸酯,在227 nm 波长处检测,其检测限分别为 2. 1 μg·mL - 1和 2. 1 μg·mL - 1,均低于氯哌斯汀芬地柞酸原料药中 GTIs 的规定限值。由于部分 GTIs 超低水平的限度要求,或者具有与 API 相同或者相似结构的发色基团,甚至缺少发色基团,使得 UV 检测器往往不能提供足够的灵敏度和选择性,需要结合衍生化的手段对样品进行前处理,使其可以适用于 UV 检测。Luo 等[28]建立了检测氯霉素中痕量 4 - 硝基苯甲醛的衍生化 HPLC - UV 分析方法,并且比较了 4 个硝基取代苯肼作为衍生化试剂的优势。结果显示 3 -硝基苯肼衍生化物可以使得待测 GTI 吸收波长明显红移到 397 nm,能最大限度地减少药物基质和衍生化试剂的干扰,提高了其灵敏度,使得检测限达到0. 009 μg·mL - 1。Wang 等[18]建立了一种衍生化法反相 HPLC - UV 测定原料药中痕量肼的方法。该方法采用 2 - 羟基 - 1 - 萘醛作为衍生化试剂,与肼反应生成具有发色基团的腙类化合物,并远离原料药的吸收波长范围,减少了基质干扰,大大提高了方法的灵敏度和选择性。同 UV 检测器一样,DAD 同样可以选用衍生化手段来提高方法灵敏度。Hou 等[29]建立了一种衍生化 HPLC - DAD 法,在温和条件下采用 2 - 硝基苯肼作为衍生化试剂,乙基二甲基胺丙基碳化二亚胺作为催化剂测定 8 个药物中 6 个卤代羧酸的含量,该方法的灵敏度、专属性、线性、回收率及耐用性试验均符合要求,检测限为 0. 02 ~ 0. 05 μg·mL - 1,为检测高温不稳定且缺少发色团的极性 GTIs提供了一种思路。
4. 2. 2 MS 检测器
HPLC-MS是一种检测范围广的普适性分析方法,MS 检测器因其具有较其他检测器更高的灵敏度和特异性的特点,常用于各类基因毒性杂质的痕量检测。Zhang 等[30]采用HPLC-MS/MS法测定氟胞嘧啶中的 N,N-二甲基苯胺,MRM 模式下对杂质进行定量分析,检测限为0. 1ng·mL - 1。Bhavani 等[31]建立了一种灵敏的LC - MS法测定阿扎那韦硫酸盐药物中的遗传毒性杂质2-[4( 吡啶-2-基) 苄基]肼羧酸酯,该方法检测限低至 0. 3 μg·g-1,回收率在 96. 4%~ 100. 4% 。Sun 等[32]在发现合成雷沙吉兰时会引入 2 种炔丙基氨基甲酸酯类结构的 GTIs,建立了 HPLC - MS 法以测定其含量,该方法具有良好的特异性、线性、精密性和准确性,其检测限为 0. 07 ng·mL - 1。Guo 等[33]利用 LC-MS 法测定 12 种磺酸盐GTIs 的含量,并对 2 种离子源 ESI 和 APCI 进行了评价。结果表明 ESI 和 APCI 得到的 MS 结果在检测限和相对丰度上都有很大的不同。在 SRM 模式下选择APCI 负离子模式可使检测限更低。在 ESI 电离条件下,GTIs 的前体离子[M + H]+ 容易被[M + NH4]+离子或[M + Na]+ 离子所竞争,导致其丰度降低,灵敏度较差。
4. 2. 3 带电气溶胶检测器和蒸发光散射检测器
带电气溶胶检测器( CAD) 和蒸发光散射检测器( ELSD) 是近年来较新颖的检测器,由于其检测原理不受样品的光学特性或者结构特点所影响,可以对不含发色基团的样品产生响应而在药物分析中得到了广泛的应用。Lecoeur 等[34]建立了 HPLC - ELSD法用于检测邻苯二甲酸二( 2 - 乙基己) 酯,并与超临界流体色谱( SFC) - ELSD 法进行了对比,结果表明HPLC - ELSD 法比 SFC - ELSD 法更加准确。Abdelrahman 等[35]建立了一种 HPLC - CAD 法用于测定醋酸氢化可的松、福西地酸中的杂质以及羟苯甲酸甲酯,结果表明该方法具有良好的系统适用性、线性、回收率、耐用性以及准确性。
4. 2. 4 超高效液相色谱法
超高效液相色谱( UPLC) 分离原理与 HPLC 一致,但 UPLC 具有耐高压且柱填料粒径更小的特点,在痕量基因毒性杂质检测中提供了一种较 HPLC 分离度更好,灵敏度更高的分析思路。Reddy 等[36]测定了佐米曲普坦中的 4 个 GTIs,由于之前所报道HPLC 法分析时间长且分离效率低,作者重新建立了一种灵敏度高、选择性好的 UPLC - MS /MS 方法,采用 3. 0 μm 粒径的 C8 柱,可以同时分离和定量 4 个GTIs,检测限降至 75 μg·g - 1之下。Wang 等[37]开发了 UPLC - UV 法和柱切换 HPLC - UV 法检测盐酸沙普格雷中间体中的环氧化物 GTIs; 对比2 种方法发现,UPLC 具有较高的灵敏度,而柱切换法可以较好地降低基质效应,柱切换 UPLC 法可以作为检测具有严重的基体效应和极低限度要求 GTIs 的一种新思路。
4. 2. 5 亲水作用色谱法
亲水作用色谱( HILIC) 分离原理与 HPLC 一致,主要用作大多数极性化合物的分离检测,例如磺酸类 GTIs。由于部分磺酸挥发性差,酸度强,因此不适用常规的 GC - MS 法,必要时需要进行复杂的样品前处理以保证达到所需的灵敏度。HILIC 法作为一种简便 的 替 代 方 法,近年来得到了广泛的关注。Huang 等[38]建立了一种简便灵敏的 HIPIC - ESI/MS方法用于测定痕量的 8 个烷基磺酸类 GTIs,该法选择含正电荷官能团的固定相以保持有机酸的稳定性,提高了流动相中乙腈的含量以增强 ESI/MS 信号强度,提高了整个方法的灵敏度。HILIC 也可以检测其他极性 GTIs。Shackman[39]评估了甘氨酸、苯丙氨酸和阿那匹韦中氮丙啶和 2 - 氯乙胺的含量,建立了一种无需衍生化且测定快速的 HILIC - MS 法。结果表明 2 种目标分析物的线性在 0. 5 ~ 10 mg·L - 1范围内良好,且灵敏度、重复性和准确性均符合要求。Jain 等[40]建立了一种定量分析达伐吡啶中 5 个潜在芳香胺 GTIs 的 HILIC - UV 法,在波长 280 nm 进行检测,5 个 GTIs 的检测限可以达到 7. 5μg·g - 1,具有良好的分离度、线性及准确性。
4. 3 气相色谱法
气相色谱( GC) 是检测具有挥发性的 GTIs 常用方法之一,相较于直接进样容易污染进样器和色谱柱,GC 可以选择性地将待测样品挥发至密闭顶空空间进样,具有减少挥发性成分的损失和基质效应,同时减少对进样器或色谱柱的污染。目前顶空分析技术主要包括静态顶空( static headspace,SHS) 分析以及动态顶空( dynamic headspace,DHS) 分析等技术。SHS 的原理是将供试品溶液置于恒温密封的顶空样品瓶中,使其中的挥发性组分逸出,在达到气液或气固分配平衡后,采集定量气相部分进入气相色谱仪进行分析,通过测定样品基质上方的气体成分来测定它们在原样品中的含量[41]。Karine 等[42]开发了一种 SHS -GC - MS 法用于检测原料药合成过程中产生的甲磺酸乙酯,该方法解决了直接进样导致的基质污染问题,并结合衍生化手段克服了甲磺酸乙酯可能会在进样口加热分解的困难。与 SHS 相比,DHS 则是通过惰性气体连续吹扫样品从而多次萃取其中的待测挥发组分,在吸附剂或冷阱中富集后再脱吸附,用载气载入色谱柱进行分析测定。由于气体的吹扫可以破坏密闭容器中气液两相的平衡,使挥发成分不断地从液相进入气相而被吹扫出来,因此几乎可把全部被测物萃取出来,具有取样量少,富集效率高,受基体干扰小等优点,一般情况下比静态顶空法灵敏度更高,适用于痕量基因毒性杂质的检测。Tore 等[43]采用 DHS- GC-MS法测定盐酸奇霉素原料药中的环氧乙烷,并对吹扫捕集过程进行了优化,其检测限较直接进样法及静态顶空法降低了约 2~ 3个数量级。除了进样方式之外,还可以通过选择合适的检测器来提高方法的灵敏度。GC 常联用的检测器包括氢火焰离子检测器、电子俘获检测器和质谱检测器等。
4. 3. 1 氢火焰离子检测器
氢火焰离子检测器( FID) 适用于有机化合物的常量到痕量分析,它的简便性使其成为检测挥发性有机分子的首选,是目前GC必备的检测器之一。由于磺酸酯类基因毒性杂质检测时易出现遇水分解的现象,具有一定挥发性的磺酸酯类GTIs 可以采用GC-FID法进行分析。Schulz 等[44]采用 HS-GC-FID 联用技术测定丙谷胺片中的痕量基因毒性杂质4 - 氯苯胺,检测出片剂中 其含量仅为9. 5 ng。Daoud 等[45]运用酸化乙醇作为衍生化试剂,建立了一种 SHS-GC-FID检测药物辅料中甲醛的分析方法,检测限为 2. 44 μg·g-1。FID 虽然是目前应用最广泛气相色谱检测器,然而对于检测极低限度 GTIs的选择性和灵敏度在一定程度上都受到限制,并且FID 对含杂原子的有机化合物响应值偏低,因此其灵敏度不一定能满足检测需求。张萌萌等[46]采用 GC分别联用 FID、电子俘获检测器和 MS 3 种检测器的方法测定甲磺酸达比加群酯中的甲磺酸乙酯和甲磺酸异丙酯,对比评价了不同检测器的灵敏度及空白干扰性并进行样品测定,结果表明 FID灵敏度不及MS检测器和 ECD,无法检测出样品中所含甲磺酸酯类 GTIs。
4. 3. 2 电子俘获检测器
电子俘获检测器( ECD) 常用于分析痕量电负性化合物,例如卤代烃、金属有机化合物、共轭双键化合物以及含磷、硫、羟基、硝基化合物等。在 GTIs 分析检测中,常用 GC - ECD 技术检测挥发性卤代烃。Skett 等[47]利用 GC - ECD 联用法检测并评估了一系列短链烷基卤化物的灵敏度,并证明待测氯化物、溴化物、碘化物的响应值随其组分的电负性增强依次增加。由此可见,ECD 检测含溴和含碘的烷烃化合物灵敏度更高,且适用于含有多个卤原子的 GTIs。除了烷基卤化物之外,ECD 也适用于卤代芳香烃以及硝基芳香烃的检测。Ho 等[48]用 HS - GC - ECD法测定了高沸点卤代烷烃、卤代芳香烃以及硝基芳香烃等一系列 GTIs,进一步扩大了 HS - GC - ECD法的适用范围及灵敏度,检测限最低可以达到 0. 0025 μg·g - 1。ECD 同样适用于检测磺酸酯类基因毒性杂质。张倩颖等[49] 采取原位衍生 化 GC - ECD法,检测甲磺酸伊马替尼中 3 个磺酸酯类 GTIs,定量限最低为 0. 0025 μg·mL - 1。虽然 ECD 泛适用于电负性化合物,然而其对不含电负性基团的成分无信号或者信号很小,并且对部分卤化物的选择性较差。
4. 3. 3 MS 检测器
GC- MS也常用于易挥发性基因毒性杂质的痕量检测,当其他检测器的灵敏度和选择性无法满足检测需 求 时,通常首先考虑选用 MS 检测器。Liu等[50]探讨了 HS- GC 联用 ECD 和 MS 检测器测定氯甲烷和氯乙烷,结果表明 2 个氯化物在色谱柱中分离度较差,并且在相同条件和浓度下,联用 ECD 无法检测出氯乙烷,但是联用 MS 检测器选择 SIM 模式检测 m /z 64 离子时,其信噪比为 109,表明在烷烃的一氯代物的检测中,HS - GC - MS 法的灵敏度和特异性明显高于 HS-GC-ECD 法。GC-MS 分析磺酸酯类 GTIs 也有较高的灵敏度。Liu 等[51] 建 立 了GC - MS /MS法测定药物中 9 个常见磺酸酯类 GTIs含量,采用 MRM 模式分别提取 9 个 GTIs 的离子对可有效降低基体效应,定量限在 0. 10 ~ 1. 05 ng·mL - 1范围内,低于其他方法( 2. 5 ~ 1 500 ng·mL-1) 。除此之外,MS 检测器在检测没有发色基团的 GTIs 上也具有优势。Chen 等[52]采用 GC - MS 法,SIM 模式下检测药物合成工艺中引入的环氧化物 GTIs 的含量。由于该药物热不稳定,作者通过优化进样口温度以降低基 质 干 扰,测得环氧化合物 GTI 的 LOD 为0. 001 4 μg·mL - 1,远低于依照 ICH M7 指南计算的限度。Sun 等[53]建立了一种原位衍生 HS - GC - MS 法用于测定原料药中的痕量肼,该方法使用丙酮或丙酮 - D6作为衍生化试剂,能够测定 1 ng·mg - 1水平的肼。MS 检测器因其高灵敏性及可广泛应用的特点,成为检测痕量 GTIs 的首选。
4. 4 毛细管电泳法
毛细管电泳( CE) 是以毛细管为分离通道,高压直流电场为驱动力的一种液相分离分析技术,可用于药物中痕量 GTIs 的富集和检测。Khan 等[54]建立了一种简便快速的 CE 法用于测定 GTIs 硫酸二甲酯( DMS) 和氯乙酰氯( CAC) 。DMS 和 CAC 在 pH 为5. 6 时均以单负离子存在,在外加电场的作用下向阳极迁移,省去了烦琐的衍生化步骤。van Wijk等[55]使用4 - 二甲氨基吡啶( DMAP) 或 1 - (4-吡啶基)哌啶-4-甲酸正丁酯( BPPC) 作为衍生化试剂,富集待测物后联用CE - MS 法测定了4个卤代烷烃GTIs。结果表明,CE-MS 预富集法的灵敏度较LC-MS法更高。虽然这2种方法都适用于GTIs 的痕量 分 析,但在这4种卤代烷烃 GTIs 检 测 中,CE-MS法更好地利用了衍生化产物的特性,提高了检测的特异性和样品的分离效率。
4. 5 超临界流体色谱法
超临界流体色谱( SFC) 是一种以超临界流体如二氧化碳等作为流动相,调节其溶剂化能力以达到分离分析目的的方法。SFC 通过改变温度和压力等参数来控制超临界流体转化为气体、液体或保持其超临界流体状态,可以联用各类 LC 或 GC 的检测器,具有分离效率高及分析时间短等优点,为理化性质各异的GTIs 分析提供新的分析思路。Schmidtsdorff等[56]建立了一种用 SFC 联用二极管阵列( photodiodearray,PDA) 检测器以及质谱检测器技术检测沙坦类药物中 10 个亚硝胺类 GTIs 的方法,PDA 扫描记录190 ~ 400 nm 范围并在 230 nm 下进行测定,MS 检测器在 SRM 模式下对待测 GTIs 进行定量分析,其检测限值在 0. 22 ~ 4. 55 ng·mL - 1范围内,灵敏度与 FDA和 EMA 发布的 LC 法和 GC - MS /MS 方法相当,但分析速度更快,并且能够一次性检测多种与 API 相关的 GTIs。Lecoeur 等[34]采用 SFC 法对多种致突变化合物和线性多环芳烃进行检测,并且与 HPLC -ELSD 法作对比,结果表明 SFC 法的灵敏度更高。
4. 6 离子色谱法
使用离子色谱( IC) 法测量带电离子如常见阴离子( 卤素离子) 或常见阳离子( 铵离子) 时较其他分析手段分析时间更少,灵敏度更好,因此烷基氯化物类和肼类 GTIs 均可考虑使用 IC 法进行检测。Harihara 等[57]建立了一种抑制电导离子色谱法检测厄贝沙坦原料药中的叠氮化合物,与 GC - MS 法和反相 LC 法相比,IC 法允许直接进样,避免了烦琐的衍生或样品处理,且 灵 敏 度 高。Karthikeyan等[58]测定盐酸司维拉姆中的丙烯胺( AAM) 含量,由于 AAM 的紫外吸收能力差,且 SPE - HS - GC 法无法萃取完全等问题,作者选择非抑制电导检测的IC 法,省去了衍生化的过程,可以直接检测质子化形式的有机胺,并利用阳离子抑制器提高了方法的灵敏度。除 此 之 外,该方法一针运行时间小于8 min,节省了检测成本。
五、总结
基因毒性杂质是近年来的研究热点。对于杂质的基因毒性评估应从药物的合成、生产及贮存过程入手,通过分析不同杂质的结构特点及理化性质判断其中可能引入的潜在的基因毒性杂质,结合数据库文献查询、QSAR 评估和遗传毒性试验对其进行分类,根据指南中明确的不同类别杂质可接受摄入量计算方法确定其合理限度,并建立起满足灵敏度、专属性、稳定性、准确性的分析方法并进行验证。
Adrian 等[59]对 H1 抗组胺药盐酸西替利嗪合成过程中可能产生潜在基因毒性杂质的风险作出了详尽的评估,包括涉及到的起始物料、溶剂、催化剂、可能产生的中间体等各项可能引入基因毒性杂质的途径。在确认这些杂质无法在过程中被完全清除且可能引入到终产品中后,作者根据其是否具有警示结构或是否有相关毒理学数据等对杂质进行分类并制定了限度。最后,作者建立了 HPLC - MS 法对 1 批盐酸西替利嗪及其杂质进行了检测,为进一步提出合理的控制策略提供支持。在 Adrian 等[59]研究中,作者依据化学知识以及对合成途径的充分理解,对可能引入的基因毒性杂质及其转入终产品的理论风险评估作出了合理的预测; 但是研究仍有不足之处:作者并未对潜在基因毒性杂质进行充分的危害性评估。在确认该潜在遗传毒性杂质可能引入到终产品之后,仅仅通过警示结构对杂质进行分类是不合理的,有可能会对杂质进行错误的分类,以至于建立的杂质限度并不合理。目前的基因毒性杂质研究中,有部分研究人员在发现了具有警示结构的杂质后便将其定性为基因毒性杂质进行控制,这样做有可能会将一般杂质误定为遗传毒性杂质,从而一味降低限度,除了把资源浪费在开发高敏感性分析方法之外,也造成了工艺开发过程中杂质控制困难,消耗不必要的成本等问题。因此,参照 ICH M7 ( R1) 以及ICH S2( R1) 等指导原则对杂质进行充分的遗传毒性危害性评估是必要的。
对杂质的基因毒性研究应不仅仅停留在确定其遗传终点和限度要求上,还应结合质量标准,考虑如何建立科学的基因毒性杂质控制策略。例如采取优化相关工艺步骤或参数,建立中间体验收标准,开发高效的纯化方法等手段去除终产品中的遗传毒性杂质或者将其降低至可接受的限度,充分理解质量源于设计( quality by design,QbD) 理念,保证和提高药品质量。
参考文献
[1] SZEKELY G,AMORES DE SOUSA MC,GIL M,et al. Genotoxic impurities in pharmaceutical manufacturing: sources,regulations,and mitigation[J]. Chem Rev,2015,115( 16) : 8182
[2] MÜLLER L,MAUTHE RJ,RILEY CM,et al. A rationale for determining,testing,and controlling specific impurities in pharmaceuticals that possess potential for genotoxicity[J]. Regul ToxicolPharmacol,2006,44( 3) : 198
[3] ICH M7( R1) . Assessment and Control of DNA Reactive ( Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk[EB /OL]. ( 2017 - 05 - 31) [2020 - 02 - 03]. https: / /database. ich. org /sites/default /files/M7_R1_Guideline. pdf
[4] USA National Library of Medicine National Institutes of Health[EB /OL]. ( 2019 - 12 - 05) [2020 - 02 - 03]. https: / /www. nlm. nih. gov /databases/download /toxlinesubset. html
[5] CUNNINGHAM AR,ROSENKRANZ HS,ZHANG YP,et al. Identification of genotoxic and non - genotoxic alerts for cancer inmice: the carcinogenic potency database[J]. Mutat Res,1998,398( 1 - 2) : 1
[6] 祝清芬,魏霞,耿雪,等 . 左羟丙哌嗪和羟苯磺酸钙中 2 个 1类杂质的遗传毒性( Q) SAR 评价及质控限度评估[J]. 药物分析杂志,2018,38( 2) : 354 ZHU QF,WEI X,GENG X,et al. Assessment and control of two class 1 genotoxic impurities in levodropropizine and calcium dobesilate[J]. Chin J Pharm Anal,2018,38( 2) : 354
[7] ICH S2( R1) . Guidance on Genotoxicity Testing and Data Inter pretation for Pharmaceuticals Intended for Human Use[EB /OL].( 2011 - 12 - 09) [2020 - 02 - 03]. https: / /database. ich. org /sites/default /files/S2%28R1%29%20Guideline. pdf
[8] TAKUMI A,KAWAMATA Y,SAKAI R,et al. In vitro and in vivo genotoxicity studies on monosodium L - glutamate monohydrate[J]. Regul Toxicol Pharmacol,2019,107: 104399
[9] European Medicines Agency. Guideline on the Limits of Genotoxic Impurities[EB/OL]. ( 2006 - 06 - 28) [2020 - 02 - 03].www. emea. europa. eu /docs/en_GB /document_library /Scientific_guideline /2009 /09 /WC500002903. pdf
[10] European Directorate for Quality and Medicines & Health Care( EDQM) . Methyl,Ethyl and Isopropyl Methanesulfonate in MethanesulfonicAcid[ EB /OL ] .[ 2020. 02. 03 ] .
https: / /www. edqm. eu /sites/default /files/publication_calendar_for_the_9th_edition_pheur. pdf
[11] CUI Y,LIU D,BIAN J,et al. Dispersive liquid - liquid microex traction with high - performance liquid chromatography for the analysis of 1,4 - benzodioxane - 6 - aldehyde in eliglustat tartrate active pharmaceutical ingredient[J]. J Pharm Biomed Anal,2020,179: 112988
[12] DURAK BY,CHORMEY DS,FIRATM,et al.Validation of ultrasonic- assisted switchable solvent liquid phase microextraction for trace determination of hormones and organochlorine pesticides by GC - MS and combination with QuEChERS[J]. Food Chem,2020,305: 125487
[13] PENG XT,ZHAO X,FENG YQ. Preparation of phenothiazinebonded silica gel as sorbents of solid phase extraction and their application for determination of nitrobenzene compounds in environmental water by gas chromatography - mass spectrometry[J]. JChromatogr A,2011,1218( 52) : 9314
[14] TZANAVARAS PD,THEMISTOKLEOUS S,ZACHARIS CK. Automated fluorimetric determination of the genotoxic impurity hydrazine in allopurinol pharmaceuticals using zone fluidics and on - line solid phase extraction[J]. J Pharm Biomed Anal,2020,177: 112887
[15] COLÓN I,RICHOLL SM . Determination of methyl and ethyl esters of methanesulfonic,benzenesulfonic and p - toluenesulfonic acids in active pharmaceutical ingredients by solid - phase micro extraction( SPME) coupled to GC/SIM - MS[J]. J Pharm Biomed Anal,2005,39( 3 - 4) : 477
[16] ZHOU J,XU J,ZHENG X,et al. Determination of methyl meth anesulfonate and ethyl methanesulfonate in methanesulfonic acid by derivatization followed by high ‐ performance liquid chromatography with ultraviolet detection[J]. J Sep Sci,2017,40( 17) : 3414
[17] LI M,GU C,LUO L,et al. Determination of trace methanesul fonates in drug matrix using derivatization and headspace single drop microextraction followed by high - performance liquid chromatography with ultraviolet detection[J]. J Chromatogr A,2019,1591: 131
[18] WANG J,YANG S,ZHANG K. A simple and sensitive method to analyze genotoxic impurity hydrazine in pharmaceutical materials[J]. J Pharm Biomed Anal,2016,126: 141
[19] CANAVESI R,APRILE S,DEL GROSSO E,et al. Semicarbaz ide hydrochloride as impurity in drug substances: a validated LC -DAD - UV method for its determination in carbazochrome and carbazochrome sodium sulfonate[J]. Chromatographia,2017,80( 10) : 1535
[20] LIU D Q,SUN M,KORD AS. Recent advances in trace analysis of pharmaceutical genotoxic impurities[J]. J Pharm Biomed Anal,2010,51( 5) : 999
[21] BAI L,SUN M,AN J,et al. Enhancing the detection sensitivity of trace analysis of pharmaceutical genotoxic impurities by chemical derivatization and coordination ion spray - mass spectrometry[J]. JChromatogr A,2010,1217( 3) : 302
[22] ZHENG X,LUO L,ZHOU J,et al. Development and validation of a generalderivatization HPLC method for the trace analysis ofacyl chlorides in lipophilic drug substances[J]. J Pharm BiomedAnal,2017,140: 327
[23] WU L,LIU DQ,VOGT FG,et al. Gas - phase derivatization via the Meerwein reaction for selective and sensitive LC - MS analysis of epoxides in active pharmaceutical ingredients[J]. J Pharm Biomed Anal,2011,56( 5) : 1106
[24] WU X,ZHU L,VISKY D,et al. Derivatization of genotoxic nitroaromatic impurities for trace analysis by LC - MS[J]. AnalMethods,2014,6( 18) : 7277
[25] VANHOENACKER G,DUMONT E,DAVID F,et al. Determination of arylamines and aminopyridines in pharmaceutical products using in - situ derivatization and liquid chromatography - massspectrometry[J]. J Chromatogr A,2009,1216( 16) : 3563
[26] GRIˇCAR M,ANDRENŠEK S. Determination of azide impurity insartans using reversed - phase HPLC with UV detection[J]. JPharm Biomed Anal,2016,125: 27
[27] GARCíA A,RUPÉREZ FJ,CEPPA F,et al. Development of chromatographic methods for the determination of genotoxic impurities in cloperastine fendizoate[J]. J Pharm Biomed Anal,2012,61: 230
[28] LUO L,GU C,LI M,et al. Determination of residual 4-nitrobenzaldehyde in chloramphenicol and its pharmaceutical for mulation by HPLC with UV /Vis detection after derivatization with 3 - nitrophenylhydrazine[J]. J Pharm Biomed Anal,2018,156: 307
[29] HOU D,FAN J,HAN L,et al. Determination of small halogenated carboxylic acid residues in drug substances by high performance liquid chromatography - diode array detection following derivatization with nitro - substituted phenylhydrazines[J]. J Chromatogr A,2016,1438: 46
[30] 张云峰,钱建钦,王建. HPLC - MS /MS 法分析氟胞嘧啶中痕量基因毒性杂 质 N,N - 二 甲 基 苯 胺[J]. 药 物 分 析 杂 志,2017,37( 2) : 265 ZHANG YF,QIAN JQ,WANG J. Determination of trace level ofgenotoxic impurity N, N - dimethylaniline in flucytosine byHPLC - MS /MS[J]. Chin J Pharm Anal,2017,37( 2) : 265
[31] SRINIVASU N,RAMACHANDRAN D. Determination of genotoxic impurity in atazanavir sulphate drug substance by LC - MS[J].J Pharm Biomed Anal,2017,132: 156
[32] SUN Y,ZHANG X,YAN Y,et al. Identification and genotoxicityevaluation of two carbamate impurities in rasagiline[J]. RSC Adv,2016,6( 108) : 106268
[33] GUO T,SHI Y,ZHENG L,et al. Rapid and simultaneous determination of sulfonate ester genotoxic impurities in drug substance by liquid chromatography coupled to tandem mass spectrometry:comparison of different ionization modes[J]. J Chromatogr A,2014,1355: 73
[34] LECOEUR M,DECAUDIN B,GUILLOTIN Y,et al. Comparison of high - performance liquid chromatography and supercritical fluid chromatography using evaporative light scattering detection for thedetermination of plasticizers in medical devices[J]. J ChromatogrA,2015,1417: 104
[35] ABDELRAHMAN MM,EMAM RA,ALI NW,et al. Novel eco -friendly chromatographic determinations of hydrocortisone acetate,
fusidic acid,their pharmacologically active impurities and pharma ceutical excipients: a comparative study[J]. Chem Pap,2020,74: 2175
[36] VIJAYA BHASKAR REDDY A,VENUGOPAL N,MADHAVI G,et al. A selective and sensitive UPLC - MS /MS approach for trace level quantification of four potential genotoxic impurities in zolmitriptan drug substance[J]. J Pharm Biomed Anal,2013,84: 84
[37] WANG R,ZHU Z,QIU X,et al. Determination of epoxide impurity in sarpogrelate hydrochloride intermediate by UHPLC and column - switching liquid chromatography[J]. J Pharm Biomed Anal,2019,174: 57
[38] HUANG Z,FRANCIS R,ZHA Y,et al. Development of a simple method for quantitation of methanesulfonic acid at low ppm level using hydrophilic interaction chromatography coupled with ESI - MS[J]. J Pharm Biomed Anal,2015,102: 17
[39] SHACKMAN JG. Rapid,direct,and sensitive determination of aziridine and 2 - chloroethylamine by hydrophilic interaction liquid chromatography - mass spectrometry[J]. MethodsX,2019,6: 2176
[40] JAIN M,SRIVASTAVA V,KUMAR R,et al. Determination of five potential genotoxic impurities in dalfampridine using liquid chromatography[J]. J Pharm Biomed Anal,2017,133: 27
[41] RODINKOV OV,BUGAICHENKO AS,MOSKVIN LN. Static headspace analysis and its current status[J]. J Anal Chem,2020,75(1) : 1
[42] JACQ K,DELANEY E,TEASDALE A,et al. Development and validation of an automated static headspace gas chromatography-mass spectrometry ( SHS - GC - MS) method for monitoring the formation of ethyl methane sulfonate from ethanol and methane sulfonic acid[J]. J Pharm Biomed Anal,2008,48( 5) : 1339
[43] RAMSTAD T,MILLER LS,THOMAS VN. Determination of residual ethylene oxide in spectinomycin hydrochloride bulk drug by dynamic headspace gas chromatography[J]. J AOAC Int,1993,76( 2) : 313
[44] SCHULZ K,OBERDIECK U,WEITSCHIES W. Degradation products of proguanil—4 - chloroaniline and related components with regard to genotoxicity[J]. Chem Pap,2013,67( 6) : 657
[45] DAOUD AGHA DIT DAOUDY B,AL - KHAYAT MA,KARA BET F,et al. A robust static headspace GC - FID method to detect and quantify formaldehyde impurity in pharmaceutical excipients[J]. J Anal Methods Chem,2018: 4526396
[46] 张萌萌,姚晓华,蒋元森,等. 不同检测器 - 顶空气相色谱法测定甲磺酸达比加群酯中的甲磺酸乙酯和甲磺酸异丙酯[J].中国医药工业杂志,2015,46( 10) : 1108 ZHANG MM,YAO XH,JIANG YS,et al. Comparison on different detectors for the determination of ethyl and isopropyl methane sulfonate in dabigatran etexilate mesylate by headspace gas chroma-tography[J]. Chin J Pharm,2015,46( 10) : 1108
[47] SKETT P W,SMITH RJ,WEBB ML. Low - level measurement of potent toxins[J]. Anal Drug Impurities,2007,1: 82
[48] HO TD,YEHL PM,CHETWYN N P,et al. Determination of trace level genotoxic impurities in small molecule drug substances using conventional headspace gas chromatography with contemporary ionicliquid diluents and electron capture detection[J]. J Chromatogr A,2014,1361: 217
[49] 张倩颖,冯芳 . GC - ECD 法检测甲磺酸伊马替尼中甲磺酸烷基酯[J]. 广州化工,2018,46(9) : 66ZHANG QY,FENG F. Determination of alkyls methanesulfonatein imatinib mesylate by GC - ECD[J]. Guangzhou Chem Ind,2018,46( 9) : 66
[50] LIU DQ,SUN M,KORD AS. Recent advances in trace analysis ofpharmaceutical genotoxic impurities[J]. J Pharm Biomed Anal,2010,51( 5) : 999
[51] LIU Z,FAN H,ZHOU Y,et al. Development and validation of a sensitive method for alkyl sulfonate genotoxic impurities determination in drug substances using gas chromatography coupled to triplequadrupole mass spectrometry[J]. J Pharm Biomed Anal,2019,168: 23
[52] CHEN L,ZHANG W,HU S. Determination of genotoxic epoxideat trace level in drug substance by direct injection GC /MS[J]. J Pharm Biomed Anal,2017,146: 103
[53] SUN M,BAI L,LIU DQ. A generic approach for the determination of trace hydrazine in drug substances using in situ derivatization - headspace GC - MS[J]. J Pharm Biomed Anal,2009,49(2) : 529
[54] KHAN M,JAYASREE K,REDDY KK,et al. A validated CEmethod for determining dimethylsulfate a carcinogen and chloroacetyl chloride a potential genotoxin at trace levels in drug substances[J]. J Pharm Biomed Anal,2012,58: 27
[55] VAN WIJK AM,NIEDERLÄNDER HAG,VAN OGTEN MD,et al.Sensitive CE - MS analysis of potentially genotoxic alkylation compounds using derivatization and electrokinetic injection[J]. Anal ChimActa,2015,874: 75
[56] SCHMIDTSDORFF S,SCHMIDT AH. Simultaneous detection ofnitrosamines and other sartan - related impurities in active pharmaceutical ingredients by supercritical fluid chromatography[J]. JPharm Biomed Anal,2019,174: 151
[57] SUBRAMANIAN NH,BABU VR,JEEVAN RG,et al. matrixelimination ion chromatography method for trace level azide determination in irbesartan drug[J]. J Chromatogr Sci,2009,47( 7) :529
[58] KARTHIKEYAN K,SHANMUGASUNDARAM P,RAMADHAS R,et al. Development and validation of rapid ion - chromatographic method with conductivity detection for trace level determination of allylamine in sevelamer drug substances[J]. J Pharm Biomed Anal,2011,54( 1) : 203
[59] LEISTNER A,HAERLING S,KREHER JD,et al. Risk assessment report of potential impurities in cetirizine dihydrochloride[J]. J Pharm Biomed Anal,2020,189: 113425