您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-11-03 23:04
医疗器械研发立项|MA策划需要考虑哪些
之所以进入医疗器械门槛高,在于高严的法规要求和不菲的前期投入。通常一个常规三类产品立项到取证上市要3年左右的时间,而是否能够成功取证是在最后注册审评阶段决定的。目前在国内没有一个非常有效的官方途径来全面评估产品注册过程的风险,企业想要顺利取证,就要有非常专业的法规技术人员来识别控制可能出现的风险。
笔者一直认为医械研发先做规划,再做事。不是简单的规划,而是“充分”的规划。运用项目管理的思维,毕竟注册不是一个人的事,而是整个团队要全力参与的,事情的返工往往是某一方面的考虑不完善导致的。注册过程的风险在产品设计开发立项时就要识别和管理。有几个方面要注意:
1.产品所要进行的认证
NMPA、CE、FDA、加拿大CMDCAS、巴西ANVISA等,不同国家、地区的认证法律法规、标准要求不同,注册流程不同。开发的产品需要考虑兼容性,项目管理的过程要考虑注册阶段的划分及每个阶段的前置任务和关联任务。
2.产品适用的法律法规和标准
重中之重,产品的性能、功能的制定除了考虑市场需求之外,其次就是考虑法规和标准的要求。查询产品适用的法律法规有哪些,包括功能、性能、材料、电气安全、生物相容性、软件、无线传输、包装、运输、安装维护、报废等方面。标准的规定是最低要求,一个质量合格的产品首先应该满足标准的规定,如产品未达到规定则检测不通过,轻则返工,重则重新设计、开模、制样。如遗漏了适用条款还要面临发补补检,时间是大大的浪费。
3.临床策略和对比产品
要不要做临床是企业非常关心的问题,项目初期策划就会考虑临床策略,是走豁免临床还是同品种比对或临床试验。原则上产品在豁免目录中可以豁免临床,但需要注意与豁免目录产品和对比产品的差异是否可以通过非临床研究证明其安全性。
同品种比对是目前难度很高的临床评价方式,可谓是”高要求条件”和“高严谨性”。表现在,使用同品种产品的临床试验数据、工艺等未公开的资料应取得授权。另外,拿江苏省来举例,同品种产品“上市多年”应为超过一个注册周期,“成熟产品”应为在中国大陆地区获准注册已经超过五个(含五个)以上的同品种产品。在《医疗器械临床评价技术指导原则》中对同品种对比临床试验数据分析过程的规定充分体现了严谨性。
临床试验的资金投入和预期用途有直接关系,多一个功能用途可能让临床费用翻一翻。临床试验的数据真实性成为了临床核查的重灾区,选一家合适的CRO和正规的临床基地显的尤为重要。所以等到产品设计完再考虑注册的问题,可能为时已晚。
附医疗器械设计开发流程图:
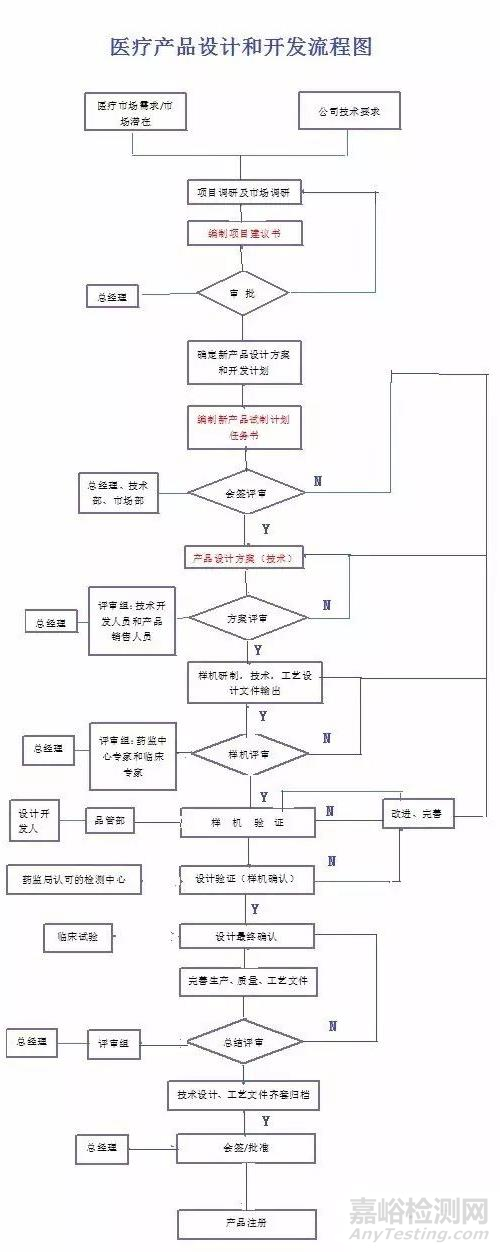

来源:Internet


