您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-03-08 04:04
摘要: 固体分散体可以有效解决难溶性药物口服生物利用度问题,但是固体分散体在储存过程中易发生相分离、重结晶等物理稳定性问题。制备工艺是影响固体分散体物理稳定性的重要因素之一。本文作者从固体分散体微观结构、宏观形态、药物与载体混合程度等角度出发,综述了制备工艺对固体分散体物理稳定性的影响机制,期望为固体分散体在药品研发中的应用提供借鉴与思考。
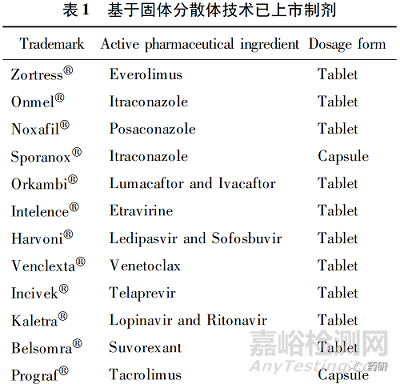
固体分散体是改善难溶性药物口服生物利用度的重要策略之一[1]。目前,全球已批准十余个基于固体分散体的难溶性药物口服制剂( 表1) 。在固体分散体中,药物大多以无序状态或单分子形式均匀分散于高分子载体中,即无定型固体分散体。与晶态药物相比,无定型固体分散体中的药物分子处于高能级态,有自发降低能级、跃迁至低能级晶体态的趋势,导致无定型固体分散体在储存过程中易出现相分离、重结晶等物理稳定性问题( 图1) [2-3],进而使药物溶出度降低,口服生物利用度降低。上述物理稳定性问题严重限制了固体分散体在改善难溶性药物口服生物利用度中的广泛应用。
南京|M4格式申报资料撰写与药品注册核查要点

近年来,已有文献综述系统阐述了固体分散体的物理稳定性与玻璃化转化温度、药物与高分子载体相容性、分子流动性、药物在高分子中的固态溶解度等因素有关[4-7]。此外,有研究显示制备工艺也是影响固体分散体物理稳定性的重要因素之一[8-9],但相关综述报道较少。因此,本文作者论述了不同固体分散体制备工艺对其物理稳定性的影响机制,为固体分散体的研究提供借鉴。
一、固体分散体制备工艺
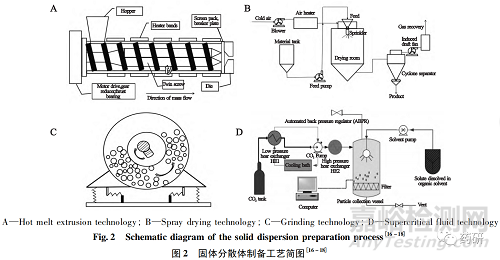
目前,已上市药品中固体分散体的制备工艺主要包括熔融法和溶剂法,其中熔融法以热熔挤出技术为主,溶剂法以喷雾干燥技术为主。热熔挤出技术是通过加热熔融、剪切混合等物理作用,使药物和载体材料在分子级别混合,冷却后形成“固态溶液”,即得无定型固体分散体。喷雾干燥技术是通过溶解破坏药物晶格结构,经干燥除去溶剂后得到无定型固体分散体[10]。除上述两种制备工艺外,科研人员还报道了研磨法、超临界流体萃取法、溶剂挥发法、旋转涂膜法等工艺[11-14]( 图2) 。采用上述不同工艺制备的固体分散体,物理稳定性差异较大,这是由于不同固体分散体制备工艺所诱导的分子相互作用和物理化学性质存在差异,从而导致固体分散体中的药物分散程度、固体分散体微观物理结构和药物晶格破坏程度等影响物理稳定性的关键因素各不相同[15]。
南京|M4格式申报资料撰写与药品注册核查要点
二、固体分散体制备工艺对其物理稳定性的影响
2. 1 固体分散体制备工艺对其微观结构的影响
在无定型固体分散体中,药物以单分子形态均匀分散于聚合物载体中,药物分子处于高能级有自发向低能级晶体态药物转化的趋势[3],这一自发过程以药物或载体分子的松弛( relaxation)运动为基础,包括单相无定型固体分散体、药物富集相、药物分子重结晶等过程( 图1) 。固体分散体物理稳定性通常用松弛来评价,它可以提供固体分散体中分子流动性信息,反映固体分散体微观结构。通常,松弛时间越短,分子迁移率越高,即分子流动性越高,重结晶速率越快。分子松弛速度可以用分子松弛时间( 分子流动性) 来描述或推算,如 Vogel-Tammann-Fulcher ( VTF) 方程:τ = τ0exp( DT0 /T-T0 ) 。其中,τ表示松弛时间,τ0表示时间常数 10-14 s,D表示与材料有关的脆性参数,T表示体系所处的温度,T0表示分子迁移率为0时的温度。该方程描述了分子松弛时间与温度的关系,可以根据松弛时间估算药物结晶速度[19]。此外,无定型物质的分子松弛过程与该物质的玻璃化转变温度( Tg ) 密切相关,当T>Tg时,松弛时间变短,分子迁移速率增加,导致药物结晶速率增加。目前,松弛研究已经成为预测无定型固体分散体物理稳定性的一种快速有效的方法,可以有效指导处方设计和制剂制备工艺选择[2]。
不同工艺制备的固体分散体,其松弛行为不同,重结晶速率亦不同。Ke等[20]分别使用研磨法、熔融法和喷雾干燥法制备固体分散体,研究了不同制备方法对药物松弛行为和物理稳定性的影响。作者采用差示扫描量热法测定样品在不同老化时间的松弛焓,将松弛焓采用Kohlrausch-Wil-liams-Watts{ φ( t) =exp[-( t /τ)β]} 方程进行拟合,获得τ( relaxation time,松弛时间) 和β( stretch power,拉伸指数,反映独立松弛状态的分布, 0<β≤1,β越小,松弛状态分布越宽) 。由于τ和β对实验误差较敏感,易导致实验结果判别不准确,该研究中,作者使用τβ ( stretched relaxation time,拉伸松弛时间) 评价松弛行为,以减小实验误差对实验结果的影响[21]。τβ值越小,表示松弛时间越短,分子迁移率越高,体系越不稳定。由 表2中松弛参数τβ可知,三种固体分散体的松弛时间由大到小依次为熔融法>喷雾干燥法>研磨法。稳定性( 室温,相对湿度75% ) 实验结果显示,采用研磨法所制备的固体分散体放置2个月时发生重结晶; 采用喷雾干燥法所制备的固体分散体放置6个月时发生重结晶; 而采用熔融法所制备的固体分散体放置6个月时仍未发生重结晶。该研究显示,固体分散体中药物重结晶速率与松弛时间呈负相关,即固体分散体中药物重结晶速率与体系分子迁移率呈正相关。Bhardwaj等[2]报道了基于溶剂法的不同制备工艺( 喷雾干燥法、冷冻干燥法和自然干燥法) 对海藻糖固体分散体物理稳定性的影响。结果同样显示,无定型固体分散体的重结晶动力学与松弛时间密切相关,三种制备工艺所制备的固体分散体松弛时间由大到小依次为: 喷雾干燥>冷冻干燥>自然干燥法; 重结晶速率与松弛时间呈现负相关,由低到高依次为: 喷雾干燥法<冷冻干燥法<自然干燥法。上述研究均表明,不同固体分散体制备工艺可显著影响其松弛行为( 分子迁移率) ,进而影响固体分散体重结晶速率。

此外,固体分散体制备工艺可以影响其药物微晶比例,进而影响物理稳定性。Karmwar 等[22]分别采用研磨法、喷雾干燥法、熔融法制备固体分散体,将样品分别放置于-20℃ ( Tg ) 条件下,观察样品重结晶现象。结果显示,研磨法所制备的固体分散体中药物未完全转变为无定型态,仍有少量微晶。三种制备工艺所制备的固体分散体重结晶速率为: 研磨法>喷雾干燥法>熔融法。这表明研磨法所制备的固体分散体中的少量药物微晶成为促使晶核生长的动力,物理稳定性较其他样品差。
另有研究表明,固体分散体制备工艺参数对其初始药物晶体比例亦有影响,进而影响物理稳定性[23]。Liu等[23]采用热熔挤出法制备吲哚美辛固体分散体,研究了挤出温度、转速等参数对固体分散体性质的影响。采用差示扫描量热法测定固体分散体中药物 晶体含量,在100℃ / 20r·min-1和110℃ /20r·min-1的挤出条件下,固体分散体中药物结晶比例分别为5. 67% 和2. 51% 。这表明随着温度升高,晶体药物比例降低,以熔融( 无定型) 状态均匀分散在高分子载体中的药物比例增加,固体分散体物理稳定性随之增加。
综上所述,固体分散体制备工艺可以影响其微观结构,如松弛行为( 分子迁移率) 、初始药物晶体比例,进而影响其物理稳定性。
2. 2 固体分散体制备工艺对其宏观形态的影响
固体分散体宏观形态主要包括粒径、颗粒形状以及颗粒表面形态。不同工艺所制备的固体分散体,其宏观形态各不相同。例如喷雾干燥法所制备的固体分散体颗粒,其粒径较小、比表面积较大、呈现多孔结构,颗粒存在明显聚集[24]。而采用热熔挤出工艺( 熔融法) 所制备的固体分散体,需经粉碎或研磨等工艺粉碎,颗粒粒径和表面形态取决于下游粉碎工艺。与喷雾干燥法相比,热熔挤出法所制备的固体分散体经粉碎后,颗粒形态不规则、表面光滑、内部结构致密无孔状结构( 图 3) [25]。上述固体分散体宏观形态上的差异可以影响固体分散体吸湿性、Tg、表面自由能等性质,进而影响物理稳定性[26]。
Agrawal 等[24]研究发现,采用热熔挤出法和喷雾干燥法两种不同方法所制备的固体分散体,在高湿条件下的吸湿性存在明显差异,物理稳定性也不同。与热熔挤出法相比,采用喷雾干燥法所制备的固体分散体颗粒内部空隙率大,在高湿条件下,吸湿性更强,吸湿性约为热熔挤出法所制备固体分散体的 2倍,物理稳定性较差。

采用同一固体分散体制备工艺,颗粒粒径不同,物理稳定性也不同。Sakurai 等[27]采用热熔挤出法制备了不同粒径的固体分散体,各颗粒间Tg( 63. 9~66. 8 ℃ ) 无明显差异。但由于固体分散体颗粒粒径越小,表面能越大,导致颗粒吸湿性增加,物理稳定性越差。
此外,固体分散体制备工艺也可以影响其Tg,进而影响物理稳定性[27]。当固体分散体颗粒粒径相似时,采用热熔挤出法所制备的固体分散体 Tg 为 63. 9 ℃,显著高于喷雾干燥法所制备的固体分散体( Tg为 42.0 ℃ ) ,物理稳定性较好。
除热熔挤出法和喷雾干燥法之外,其他固体分散体制备工艺也可以影响其宏观形态。Badens 等[28]研究了超临界流体法和喷雾冷冻干燥法对固体分散体颗粒形态的影响。喷雾冷冻干燥法所制备的固体分散体颗粒比表面积远大于超临界流体法,这种表面能差异可以显著影响物理稳定性。此外,Adeli 等[18]使用超临界流体法和溶剂蒸发法制备固体分散体。结果显示,超临界流体法制备的固体分散体为球形颗粒,粒度分布均匀; 而溶剂蒸发法制备的固体分散体为细小块状微粒,存在明显聚集( 图4) 。由于超临界流体法所制备的固体分散体结构较为致密,其结晶度小于溶剂蒸发样品,呈现出优良的体外溶出和物理稳定性。

2. 3 固体分散体制备工艺对其含水量的影响
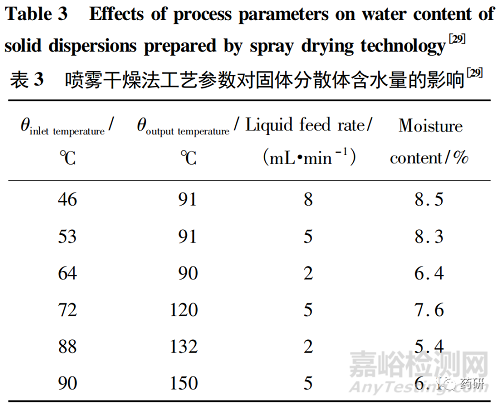
有研究表明,喷雾干燥法工艺参数,例如喷雾干燥温度、供液速度和干燥空气流速等,可以影响固体分散体颗粒的含水量,进而影响物理稳定性[29-30]。
Maa等[29]研究了喷雾干燥过程中出口温度对固体分散体性质的影响。结果表明,随着出口温度的降低,固体分散体颗粒形状由不规则的凹陷球形转变为光滑的规则球形,固体分散体颗粒含水量也不同( 表3) 。通常固体分散体含水量越高,药物重结晶速率越快。表3显示,随着出口温度的升高,固体分散体颗粒含水量逐渐降低; 但当出口温度过高时,固体分散体颗粒含水量反而增加。因此,适宜的出口温度是喷雾干燥法制备固体分散体的重要参数之一。
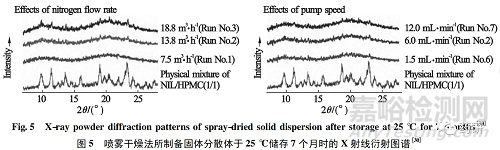
此外,Kojima 等[30]研究了喷雾干燥过程中氮气流速、供液速度对固体分散体物理稳定性的影响。根据 X 射线衍射图谱研究结果,不同工艺参数条件下,所有新制备的固体分散体中药物均为无定型态; 当样品于25℃ 密封放置7个月时,高氮气流速制备的样品出现微小的衍射峰( 图5) ,即出现重结晶; 而低氮气流速制备的样品则无明显衍射峰。不同供液泵速度制备的样品均无明显衍射峰。上述结果表明氮气流速对固体分散体物理稳定性影响较大,而供液泵速度影响较小。
2. 4 固体分散体制备工艺对药物与高分子混合程度的影响
不同制备工艺由于工艺原理不同,可以显著影响药物和载体分子间的混合程度,进而影响固体分散体内的物相分布、药物和载体分子间的相容性,导致物理稳定性不同[31]。
Yang 等[3]研究了溶剂法( 旋转薄膜包衣法)与熔融法( 热熔挤出法) 两种制备工艺对固体分散体重结晶的影响。研究结果显示,旋转薄膜包衣法所制备固体分散体重结晶速率更低,物理稳定性较好。作者推测出现这一结果的原因是旋转蒸发技术和热熔挤出技术对药物与高分子载体的混合程度有区别: 与热熔挤出技术相比,对于旋转蒸发技术,药物和高分子以溶解状态存在于溶液体系中,混合程度更高,其物理稳定性也更高。
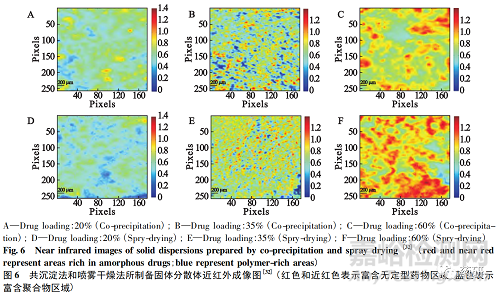
Ma等[32]使用共沉淀法和喷雾干燥法分别制备了不同载药量固体分散体,并用近红外化学成像技术分析药物和高分子载体的混合程度。如图6所示。图像中红色和近红色表示富含无定型药物区域,蓝色表示富含聚合物区域。采用共沉淀法和喷雾干燥法制备固体分散体,当载药量为20%和35% 时,药物在高分子载体中分布更均匀。当载药量相同时( 尤其是35% ) ,喷雾干燥法所制备的固体分散体中药物与高分子载体混合程度更高。表明固体分散体制备工艺可以显著影响药物与高分子载体的混合程度,进而影响其物理稳定性。
三、结论
固体分散体可以显著提高难溶性药物溶出度,改善其口服生物利用度。但是固体分散体中药物呈非晶态,与结晶态相比热力学不稳定,贮存过程中普遍存在相分离、重结晶等物理稳定性问题,最终导致溶出速率下降,生物利用度降低,这也是限制固体分散体广泛应用的主要原因之一。因此,开发基于固体分散体的口服制剂时,必须通过制备工艺选择、工艺参数优化、处方组成优化,防止产品在制备和储存过程中发生药物重结晶。其中,制备工艺是影响固体分散体物理稳定性的重要因素之一,通过研究不同固体分散体制备工艺对其物理稳定性的影响机制,可以为制备工艺的选择提供依据。目前固体分散体制备工艺对其物理稳定性的影响机制尚不完全明确,仍需进一步深入研究,以期为固体分散体的广泛应用提供理论支持。

来源:Internet


