您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-04-07 14:05
质量守恒定律是指一个系统质量的改变总是等于该系统输入和输出质量的差值;它表明质量只会从一种物质转移到另一种物质上,总量保持不变。
质量守恒主要应用于物理方面(解决热学问题以及功能转换)和化学方面(化学反应中,参加反应前各物质的质量总和等于反应后生成各物质的质量总和等)。
药品强降解试验中的质量平衡是通过样品破坏后,主成分的测量值及所有杂质的报告值之和与未破坏样品初始值接近100%的程度来确定。
强降解试验中质量平衡的目的:1)了解药物的降解路径及分子内稳定性;2)建立稳定性指示分析方法,使其适用于样品检测;3)为药品的处方、工艺、包装、贮藏条件的确定提供有益支持。
为了实现质量平衡,鉴定原料药和制剂样品中的所有杂质,几乎是不现实的(ICHQ3A仅要求鉴定超过鉴定水平的杂质)。特别是样品中含有大量未知杂质且其响应因子未知时,有时会导致原料药样品中报告的总杂含量与样品总杂含量之间存在明显差异。ICHQ3A(R2)中的要求也会产生一些差异,即只有超过原料药报告水平(RTh)的杂质包含在总杂的报告中,所有大于等于定量限(QL)的杂质都包含在计算中。制剂样品中报告的总杂含量和样品中杂质的总量之间也可能出现其他差异。产生这些差异的主要原因是原料药的报告水平小于制剂的报告水平。但并不意味着制剂样品初始稳定性(0天)中报告的总杂含量与用于生产制剂产品的原料药中(批次相同)的总杂含量相同,因为ICHQ3B(R2)仅要求报告降解产物,且仅要求报告的降解产物应包含在总杂中。ICH对杂质结果报告和数据记录的要求可以归纳为以下几点:
A、可报告的结果
1、应报告超过报告水平(RTh)的杂质含量
2、报告的数值应包含在总杂中
3、小于等于报告水平的杂质应报告为不超过报告水平(RTh)
B、记录的数据
1、应记录大于等于定量限(QL)的杂质含量,并将其包含在总杂中。
2、可记录大于等于检测限(DL)但小于定量限(QL)的杂质含量,并可记录在总杂中(应在处理记录数据和报告结果的标准操作程序中定义一致的策略)。
3、小数点右边的所有记录值均应保留在经过验证的电子表格中并包含在总杂中,然后四舍五入。
例如,表1.1显示了应如何记录和报告MDD为50mg的药物的杂质数据和相应结果,在原料药(DS)中样品含有13种杂质,其中5种是降解产物,应对其进行监测并在制剂(DP)样品中报告。假设图1.1中的色谱图既代表了DP的零天时间样品,又代表了用于制备DP的DS批次,并且在DP的生产过程中没有降解,则DS和DP中的杂质浓度将相同。表1.1显示了基于面积归一化法的质量平衡,杂质的总量为2.1818%。DS中杂质的含量降低到2.04%,因为总杂中不包括不超过报告水平(RTh)的三种杂质。DP中杂质的总杂含量仅为0.3%,因为不能报告DS中的杂质,并且仅降解物3的含量超过了0.1%(RTh)。
表1.1记录和报告MDD为50mg的药物的杂质数据和相应结果
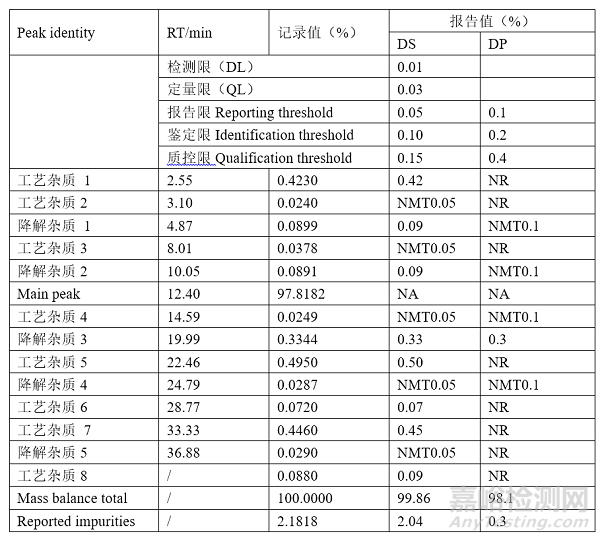
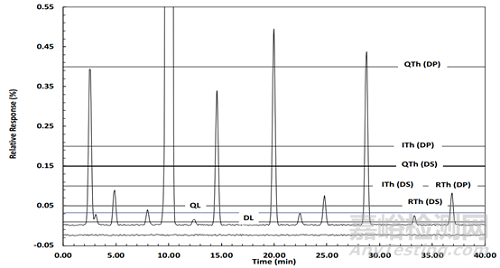
图1.1 不同浓度的杂质模拟的色谱图
质量守恒计算方法:1)主成分峰面积在线性范围内,可将供试品溶液的主峰峰面积和校正后的杂质峰面积之和除以破坏后的溶液主峰峰面积和校正后的杂质峰面积之和;2)主成分不在线性范围内,测定供试品溶液和破坏后的溶液的主成分含量和有关物质含量,计算破坏前总含量与破坏后总含量,此方法同样适用于第一种情况。
通常样品的降解量为5%-20%,药物降解至其原始量的90%为最优。如果降解条件过于剧烈,有可能发生二次降解,而在实际稳定性研究中不太可能产生二次降解产物。
影响质量守恒的因素:(1)酸/碱水解药物与水溶液中的氢离子或者氢氧根离子发生反应,生成酸性或者碱性产物。水解反应的发生需克服水解基团的水解活化能,即需要考虑水解基团所处的电子和空间位阻效应。如药物分子中含有的羧酸酯、羧酸酰胺及磷酸酰胺等基团通常水解活化能较低,是比较容易水解的位点;如醚及磺酰胺基团,一般水解活化能较高,通常不易发生水解[1]。水解速度的快慢受酸碱浓度、温度、水解时间的影响,具体的考察条件根据药物性质进行设计。
(2)光照降解光照强制降解试验的条件设置在ICHQ1B中有明确规定,可分别以固体、溶液或混悬液状态进行考察,总照度不低于1.2×106Lux·hr(冷白光灯),近紫外能量不低于200w·h/m2。应考虑样品的物理性质,并应采取措施如冷藏或置密闭容器中,以确保物理状态变化(如升华、蒸发、熔化)所造成的影响最小[2]。
(3)高温降解对于高温破坏,一般遵循阿伦尼乌斯方程,即随温度升高降解速率加快。以固体或溶液状态进行考察,通过设置较高的降解温度以在较短的时间内获得药品的降解信息。具体的考察温度和时间需结合合成工艺确定。
(4)氧化降解药物氧化降解途径主要包括2种[3],第一种是经自由基链引发的氧化,如氧浓度、温度、pH、金属离子、光照和其他可能的自由基引发物等。制剂辅料(如聚维酮、羟丙基纤维素、交联聚乙烯吡络烷酮、聚乙二醇等)中的某些杂质,如醛类、金属离子、过氧化物类,可作为氧化反应的引发剂,引发自由基链反应导致制剂发生氧化降解[4]。第二种氧化途径是通过有机过氧化物或过氧化氢氧化,该途径受有机过氧化物或过氧化氢浓度的影响。具体的考察条件应根据药物性质进行设计。
强降解试验质量守恒的可接受标准一般为90%-110%,误差主要来源于杂质校正因子、仪器误差、积分误差、操作误差;为了收紧限度,一般设为95%-105%。
如果降解试验的质量平衡不符合接受标准,可能的原因有:挥发性杂质的潜在损失;特定的降解产物未被检出(如;不保留杂质,强保留杂质);二次降解;非紫外吸收化合物的形成等。因此,在强制降解试验前需考虑样品的理化质,了解药物在各个条件下可能的降解机制以及降解反应的难易程度,合理的设计试验,以确定合适的降解条件。
参考文献
[1] MINLI. Organic chemistry of drug degradation [M].RSC Publishing,2012.
[2]ICH Q1B指导原则
[3] 马骏威, 刘涓, 刘永辉, 等. 强制降解试验在药物研发中的应用[J]. 中国现代应用药学, 2020, 37(14): 1778-1782.
[4] HARTAUERK J, ARBUTHNOT G N, BAERTSCHI S W, et al. Influence of peroxide impurities inpovidone and crospovidone on the stability of raloxifene hydrochloride intablets: Identification and control of an oxidative degradation product [J].Pharm Dev Technol, 2000, 5(3): 303-310.

来源:药事纵横


