您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-01-13 09:18
1月8日,中国医药包装协会发布了《药包材等同性/可替代性评价指南》征求意见稿,本指南是基于质量风险管理理念,对药包材(药品包装系统和组件)的等同性/可替代性(以下简称为等同性)进行评价。
药包材等同性评价指南主要用于指导药品批准文号/登记号持有人(药品上市许可持有人、药品生产企业)对已上市药品发生包装材料和容器的变更,或药包材生产企业在生产制造过程中发生的技术类变更进行评估,以验证和确认变更前后药包材产品的质量等同性。
药包材等同性/可替代性评价指南
1、适用范围
本指南是基于质量风险管理理念,对药包材(药品包装系统和组件)的等同性/可替代性(以下简称为等同性)进行评价。药包材等同性评价指南主要用于指导药品批准文号/登记号持有人(药品上市许可持有人、药品生产企业)对已上市药品发生包装材料和容器的变更【1】,或药包材生产企业在生产制造过程中发生的技术类变更【2】进行评估,以验证和确认变更前后药包材产品的质量等同性。
本指南预期用于药包材变更相关的技术性评价。作为一种技术工具,指南中给出的成对比较及等同性判定原则,也可为药包材相关的其它成对比较研究提供参考,如药品一致性评价中围绕药包材对药品质量一致性的影响所进行的相关研究。
2、名词术语
2.1药包材等同性:已上市药品发生包装材料和容器的变更或药包材生产企业在生产制造过程中发生技术类变更时,变更前后的药包材的质量属性一致性,即风险可接受程度一致性。药包材的质量属性包括安全性、保护性、功能性、相容性,即药包材的适用性。
注1:等同性并不意味着一定是相同,等同性比较通常是在预先设定的比较标准范围内进行比较。即当变更后性能满足预先设定的标准范围时,则视为具有等同性。
2.2技术类变更:药包材的技术类变更系指需要开展研究和验证,以便于后续对药品的影响进行风险评
估的变更类型。本指南中药包材的技术类变更包括:药包材的生产场地变更、药包材原材料及配方变更、药包材生产工艺和过程控制变更、药包材的包装变更,以及其它可能影响药包材质量及预期适用性的变更。
2.3高风险药包材:一般包括用于吸入制剂、注射剂、眼用制剂的药包材;新材料、新结构、新用途的药包材;国家食品药品监督管理总局根据监测数据特别要求监管的药包材。
2.4化学等同性:基于提取和/或浸出研究,药包材变更后,或其它成对比较研究中,提取物和/或浸出物谱未出现诸如提取物/浸出物数量和溶出量的增加。
2.5毒理学等同性:基于提取和/或浸出研究,药包材变更后,或其它成对比较研究中,对于化学不等同的提取和/或浸出物谱中的化合物,经过适宜的毒理学评估,其毒理学风险可以接受。
2.6药包材的生物学等同性:基于药包材的化学等同性或毒理学等同性评估,其评估结果用于豁免所需的生物学实验。
2.7药包材安全性评价:药包材安全性评价包括来自包装系统浸出物/迁移物及其相关的毒理评估;材
料/组件的体内、体外生物学评价;恰当引用食品领域法规认可的添加剂用于口服固体及液体制剂的安全性评价;如有临床证据,来自临床未跟踪到包装组件相关不良反应的证据和结论等。
3、总则
药包材等同性评价是基于风险管理理念,为减少不必要的实验,对已上市药品包装材料和容器的变更或药包材的技术类变更所进行的评价。药包材等同性评价方案宜首先考虑相关法规性文件、技术指南/指导原则、标准等要求。其次,根据风险管理理念,采用相关风险管理标准和工具,经风险评估所形成的制造商内部变更管理文件也是等同性研究的重要参考。
本指南所描述的等同性评价,是药包材变更评价或实验研究的一种技术工具。然而,对于某些变更事项,来自供应商与客户协议等的一些特定主观性评价,可能成为进行药包材等同性评价前的一个重要流程。如某些产品外观或形制的变化,在进行技术性评价之前即进行评估并达成一致后,才进入等同性评价程序。
除此之外,对于材料类相关的变更,材料自身的一些合规性测试或评估,如拟变更材料或组件符合药用要求的证据等,也是进入等同性评价程序之前的一个重要考量。
3.1药包材等同性研究的基本原则
药包材等同性研究的基本原则可包括:
1)对于药包材的任何变更,首先需识别与拟变更事项相关的风险。在风险识别基础上确认需要开展的评估内容。
2)药包材的等同性研究,作为相关法规性文件、技术指南/指导原则、标准等的补充、技术工具性文件,所描述的技术路线预期用于评估药包材适用性(安全性、保护性、功能性、相容性)相关的质量一致性。即可接受程度满足预期目的。
3)基于风险管理理念,在符合法定标准或登记标准的前提下,进行变更前后产品适用性的成对比较。成对比较通常需要预先设定可接受的标准和范围,标准和范围的设定,宜充分考虑已批准的产品质量标准、制造商内部质量控制标准、以及满足所包装药物临床需要的评价结果等。
4)充分利用国内外已达成共识并认可的现代实验技术和评价理念,如提取物/浸出物研究技术及其毒理学风险评估理念、风险管理框架下的生物学评价理念等,预期在科学评价和风险管理的基础上,减少重复性和不必要的实验。
5)在药品质量风险管理活动中,作为技术工具,其它可基于上述成对比较原则,所需进行的与药包材相关的药品质量等同性评估。
3.2药包材等同性评价的主要内容
适用时,药包材等同性评价的主要内容可包括:
1)基于风险识别和/或法规性文件所确认的关键保护性能的等同性评价;
2)基于风险识别和/或法规性文件所确认的关键功能性的等同性评价;
3)基于风险识别和/或法规性文件所确认,其他适用性能的评价,如材料相关的重要特性评价;
4)基于风险识别和/或法规性文件所确认,变更导致可能需要重新考虑生物学评价,当所变更药包材主体材料类别一致时(如均为塑料、橡胶、玻璃、金属类),生物学性能的等同性评价;
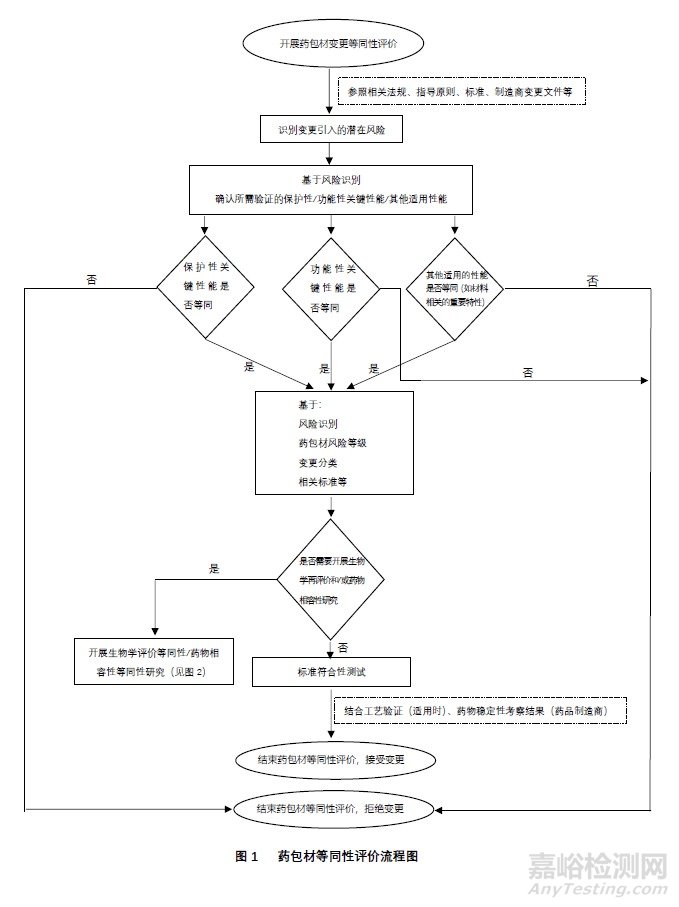
5)基于风险识别和/或法规性文件所确认,变更导致需要重新考虑包装与药物相容性研究或提取研究时,当所变更药包材主体材料类别一致时(如均为塑料、橡胶、玻璃、金属类),可提取物和/或浸出物谱的等同性评价;附录二 提供了药包材等同性评价可供参考的流程图。
4、药包材的等同性评价
药包材的等同性评价做为一种技术工具,应用于通过风险识别和/或相关法规性文件所确认的研究验证。这些研究验证,适用时,一般包括如上所述的药包材保护性关键性能的等同性评价、功能性关键性能的等同性评价、其它适用的质量特性等同性、生物学评价等同性、相容性相关等同性评价等。
4.1药包材保护性能的等同评价与判定原则
药包材的保护性一般指包装系统或包装组件,为确保制剂有效期内的质量而提供的充分保护。与药包材相关的导致制剂有效期质量下降的因素通常包括:光照、溶剂损失、活性气体接触(如氧气)、外来微生物侵入等。
药包材等同性研究中,相对应的保护性关键性能可包括:包装系统的完整性、包装系统或组件的阻隔性能、避光药物包装的避光性能等。关键性能的识别和确认应根据所变更事项的相关性确定。
通常大部分保护性能已列入药包材质量标准中,并且在药包材实施变更时需进行相关测试。标准符合性指标是保护性能等同判定的基本原则。尽管如此,变更研究中,识别保护性关键性能,在标准符合性的基础上,并采用成对比较的原则,对验证结果的比较,将有助于进一步了解变更前后这些关键性能的差异或变化趋势。其次,适用时,经论述、评估并考虑满足所包装药物临床需要的制造商内部质量控制标准,也是等同性判定标准之一。
4.2药包材功能性等同评价与判定原则
药包材的功能性通常是指按照其设计方式发挥作用的能力。传统的功能性一般多为药物递送和使用过程中,为实现其预期目的而进行的评价。除此之外,药包材的功能性还将包括某些为满足特定预期目的而进行的特殊设计。如多室袋包装、功能性吸附包装、防儿童开启包装、防重复使用的自毁式包装设计等。
药包材等同性研究中,相对应的功能性关键性能可包括:给药剂量准确性相关性能、耐穿刺性能、使用过程相关的密封性能等。对于特殊设计的药包材,还应考虑满足其设计预期的关键性能,如多室袋的虚焊相关性能等。如上所述,通常这些性能已列入药包材质量标准中,并且在药包材实施变更时需要进行测试。关键性能的识别和验证应根据所变更事项的相关性确定。
标准符合性指标是功能性等同判定的基本原则。同样,变更研究中,识别这些关键性能,在标准符合性的基础上,对验证结果的比较,将有助于进一步了解变更前后这些关键性能的差异或变化趋势。同样,适用时,经论述、评估并考虑满足所包装药物临床需要的制造商内部质量控制标准,也是等同性判定标准之一。
4.3其它适用的质量特性等同性评价和判定原则
对于某些材料的包装组件,变更可能触发与材料特性或工艺密切相关的评价,如玻璃包装的耐水性评价、金属类包装的耐腐蚀性评价、涂层的牢固度评价等等。
标准符合性是该类质量特性等同性判定的基本原则。若该类质量特性未列入产品质量标准范围,则应符合经论述的接受标准。在上述标准符合性的基础上,对验证结果的成对比较,将有助于进一步了解变更前后这些关键性能的差异或变化趋势。适用时,经论述、评估并考虑满足所包装药物临床需要的制造商内部质量控制标准,也是等同性判定标准之一。
4.4药包材安全性相关的等同性评价及判定原则
当变更事项触发需要进行药包材安全性相关评价时,所需开展的评价内容通常包括:材料/组件的体内、体外生物学评价;来自包装系统浸出物/迁移物及其相关的毒理评估;
与药包材安全性相关的等同性评价,在进入实验研究前,宜首先收集变更事项与生物学评价、相容性研究相关的技术信息,同时收集和评估历史研究数据,若经评估论述表明不需开展新的实验,则可以形成等同性研究评估报告,并确认接受变更。
注2:对于大多数非高风险药包材,若药物配方相对于所采用的包装,不具有特殊性,如酸碱性、脂溶性等,可能不需要开展与相容性/生物学评价相关的等同性研究。
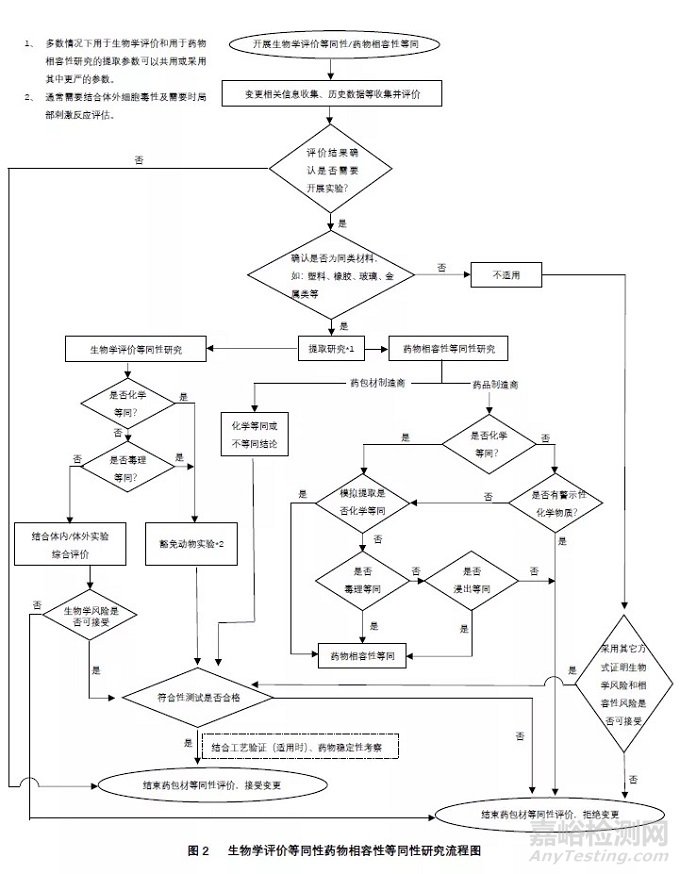
4.4.1药包材生物学等同性评价方法及判定原则
药包材的生物学评价是药包材安全性评价的重要组成部分。某些药包材的变更事项可能涉及需要重新考虑生物学评价或实验。值得注意的是,在生物学评价实践中,为减少动物实验,生物学等同性评价是一项重要应用技术之一。
药包材的生物学评价或实验可参照的标准包括:USP<87>体外生物反应试验、USP<88>体内生
物反应试验,ISO 10993(GB 16886)医疗器械生物学评价系列标准、中国药典2020版相关通则/指导原则等。通常高分子材料的高风险药包材需要根据所包装药品的临床接触性质,考虑适宜的生物学评价终点。同时,根据所包装药品的处方特性、工艺特性等,选择适宜的提取溶剂和提取参数,进行包装材料或组件的生物学评价或实验。
在进行药包材变更评价时,药包材生物学等同性评价步骤包括:
1)在收集相关信息的基础上,采用药包材材料或组件的可提取物谱比较的方式,评价变更前后的化学等同性。
2)在化学等同的前提下,结合体外细胞毒性试验,考虑豁免所需的生物学实验。
3)对于不能判定为化学等同的提取物谱,则对非等同的可提取物进行定性定量分析。
4)若可行,结合拟包装药物临床使用特性,进行与之相关的毒理学风险评估,以判定毒理学等同。
某些变更,如药包材生产企业的技术类变更,由于多数高风险药包材在实际使用中,可能用于包装不同处方特性的药品,且由于药品临床应用的多样性,导致无法根据具体药物的临床使用特性,尤其是剂量特性,进行提取物的毒理学风险评估,则对非化学等同的提取物可采用最差情况的假设,如假设化合物在论述条件下的提取量在24h内全部释放进入人体进行毒理学等同性评估。或由药品制造商关联所包装药品的临床接触特性进行评估。
生物学等同性评价作为一个可供选择用于预估生物学风险的工具,以减少不必要的动物实验,一般多用于高风险药包材的评估。
注3:对于玻璃,即使是高风险品种,传统意义上,一般不采用生物学实验或评价方式进行安全性评估。而对于金属材料,经选择和验证的金属材料牌号及其加工和预期使用过程中耐腐蚀性可能是重点需要关注的。但对于不具有相同或更高风险级别接触途径安全应用史的新材料、新工艺、新涂层产品,若需要进行评价时,可参照进行。
化学等同性和毒理学等同性判定原则见“附录一化学等同性和毒理学等同性判定原则”。
4.4.2药包材与药物相容性研究的等同性评价及判定原则
药品包装与药物相容性研究可参照的指导原则包括:国家药监局发布的“化学药品注射剂与塑料包装材料相容性研究技术指导原则“、“化学药品注射剂与药用玻璃包装容器相容性研究技术指导原则”、“化学药品与弹性体密封件相容性研究技术指导原则”。USP<1663>“药用包装/给药系统相关可提取物的评估”、USP<1664>“药用包装/给药系统相关浸出物的评估”。在上述指导原则中,药包材的相容性研究均包括可提取物研究、浸出物研究,同时,提取物和浸出物研究结果均需进行相应的毒理学风险评估从而实现与相容性相关的安全性评价。
药包材的部分变更事项,可能会触发药包材企业在相容性研究方面的风险再评估,尤其是提取物
谱的再评估。药物相容性研究等同性评价步骤包括:
1)通常对于高风险药包材,可行时,根据拟包装药物的处方、工艺特性,选择适宜的提取溶剂和提取参数,对药包材材料或组件的可提取物谱进行成对比较,评价变更前后提取物谱的化学等同性。
2)在化学等同的前提下,结合体外细胞毒性试验,考虑豁免所需的可提取物毒理学风险评估。
3)对于药品生产企业,在提取物谱等同性研究过程中,可行时,开展模拟提取物谱的等同性研究,并判定变更前后的化学等同性。
4)对于不能判定为化学等同的提取物谱/模拟提取物谱,则对非等同的可提取物进行定性定量分析。
5)若可行,结合拟包装药物临床使用特性,进行与之相关的毒理学风险评估,以判定毒理学等同性。
同样,某些情况下,如药包材生产企业的技术类变更,由于多数高风险药包材在实际使用中,可能用于包装不同处方特性的药品,且由于药品临床应用的多样性,导致无法根据具体药物的临床使用特性,尤其是剂量特性,进行提取物的毒理学风险评估。需要时,可对非化学等同的提取物采用最差情况的假设,如假设化合物在论述条件下的提取量在24h内全部释放进入人体进行毒理学等同性评估。或由药品制造商关联所包装药品的临床接触特性进行评估。
6)对于药品制造商,若上述提取研究结果不能判定为毒理学等同,则可进一步进行浸出物及其毒理学评估,以判定在实际包装药物浸出物的风险是否可以接受。
化学等同性和毒理学等同性判定原则见“附录一化学等同性和毒理学等同性判定原则”。
4.5变更后药包材产品的标准符合性测试
通过上述适用的等同性评价和其它所需的变更研究,对拟变更的药包材根据所批准的标准开展标准符合性测试。某些情况下的变更事项,可能同时涉及药包材质量标准的变更,则所采用的拟变更质量标准应经过论述和验证。
5、药包材等同性评价的结果及应用
药包材等同性评价是已上市药品发生包装材料和容器的变更或药包材生产企业在生产制造过程中发生的技术类变更时,所开展的变更评价的重要组成部分。本指南所述的等同性评价,可能并不是某些药包材变更事项所需评价内容的全部,因而,本指南所形成的评价/研究结果,结合其它研究结果,尤其是充分考虑满足风险管理要求和满足临床预期,用于支持是否接受与药包材相关的拟变更事项。
实践当中,需要结合相关技术类法规要求和/或经风险识别确认的其它验证项目,如适用时,结合所需的工艺验证结果、所包装药物的稳定性验证结果等,完成最终的变更评价,并得出评价是否被接受的结论。
附录一 化学等同性和毒理学等同性判定原则
1、化学等同性一般判定原则
对拟变更或成对比较的样品采用经论述的相同的提取条件提取,对于大于分析评估阈值(AET)的峰比较提取物谱差异,变更后样品提取物谱中不应出现多余峰,且同时不应出现峰面积增高[注2]的峰。
注4:化合物峰面积的轻微增高通过适宜的论述,也可以认定为该化合物为化学等同。如一般通过对所采用半定量分析方法变异性的统计学评估,结合化合物峰面积轻微增高的程度进行论述。
2、毒理学等同性一般判定原则
2.1按上述原则,经评估确认为具有化学等同性的药包材,同时认为具有毒理学等同性。
2.2按上述原则所述,经评估确认为不具有化学等同性的药包材,对识别出的非化学等同提取物/浸出物,按相关指导原则或标准进行毒理学风险评估,评估结果毒理学风险可接受,则认为具有毒理学等同性[注3]。
注5:当化学不等同时,毒理学评估表明化合物可能具有局部生物学反应,如刺激作用,根据相关标准或指导原则,结合药包材临床接触性质判定需要考察刺激反应(如眼用制剂、破损皮肤、粘膜接触的软膏类制剂等),或者通过毒理学风险评估应导出刺激性可接受限量,否则,宜进行所需的刺激实验。
化学等同性和毒理学等同性评价除用于豁免不必要的动物实验和减少相容性研究再评价的定性定
量、毒理学评估工作量外,作为一种可行的技术工具,还可在变更评价早期预测安全性相关风险。化学等同性和毒理学等同性的研究和判定均须有足够经验的化学家和毒理学家完成。
参考文献
【1】 CDE,已上市化学药品药学变更研究技术指导原则(征求意见稿)
【2】 T/CNPPA 3009-2020 药包材变更研究技术指南
【3】 Guidance for Industry Changes to an Approved NDA or ANDA, FDA,2004
【4】 Biological evaluation of medical devices —Part 18:Chemical characterization of medical device materials within a risk management process
【5】 化学药品注射剂与塑料包装材料相容性研究技术指导原则
【6】 化学药品注射剂与药用玻璃包装容器相容性研究技术指导原则
【7】 化学药品与弹性体密封件相容性研究技术指导原则
【8】 USP <1663> 药用包装/给药系统相关可提取物的评估
【9】 USP <1664> 药用包装/给药系统相关浸出物的评估
【10】 ISO 10993.1-2018 Biological evaluation of medical devices —Part 1: Evaluation and testing within a risk management process

来源:中国医药包装协会


