您当前的位置:检测资讯 > 实验管理
嘉峪检测网 2020-09-13 16:39
样品溶剂的选择要求,先简单的概括一下:
1、能够溶解待测物质;
2、不影响待测物质检测;
3、能够在一定时间范围内保持待测物质稳定。
什么是溶剂效应?
样品溶液的溶剂强度强于流动相溶剂强度时可能会造成的峰展宽、峰分叉现象。即色谱图上较早洗脱的峰前沿或者开叉,与此同时较晚洗脱的峰则较为正常的现象。这些现象可能是由以下原因引起的——样品溶液的溶剂很可能强于流动相。
可能发生溶剂效应的情况:出峰时间早;保留弱;进样量大
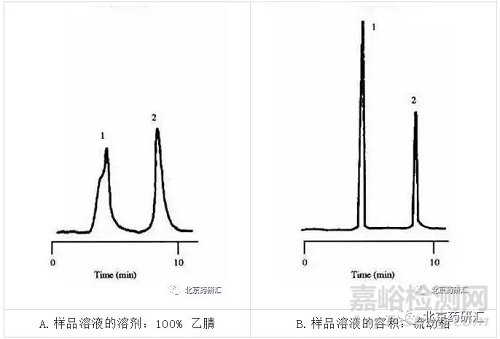
上图中,色谱图上较早洗脱的峰扭曲变形或者开叉,与此同时较晚洗脱的峰则较为尖锐与对称,这些现象显示一个比较特殊的起因――样品溶液的溶剂很可能强于流动相。此种强溶剂效应的例子在左图中可见。此处的样 品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水。第一个峰是开叉的,并且与第二个峰相比,明显地变宽 了。当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐。见右图。
为什么呢?这是因为当样品进样时,有可能出现峰展宽,最佳的样品溶液组成和体积将会保持在10%甚至更低,在这个例子里,当样品溶剂与流动相溶剂强度不同时,换句话来说,也就是样品未用流动相溶解,因此,有些样品分子溶解在强溶剂中,并随强溶剂流过柱子,而有些则溶解在流动相中,从而导致峰分叉。
当样品与流动相强度相差较小,进样影响也会小,第一个峰可能会宽于第二个峰,而当这种展宽导致必要的分离度降低时,这样情况应引起注意,在左图中,使用一根短柱,和5μL进样,这与最佳进样体积4μL相近,用了极性更强的溶剂导致分离度明显的降低,从2.1降到1.5(如右图,分离度为2或更大是评估一个完善方法的一个必要参数,也是每个方法的验证参数,1.5只是一个基本的分离度,任何一个方法或一根柱子都必需满足这个条件,当进样为一倍时,也就是10μL时,分离度更一步降低,此方法就不行了。
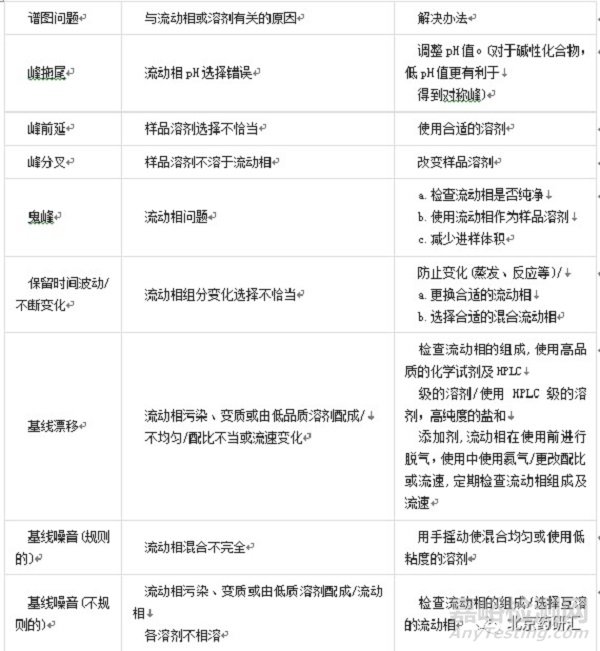
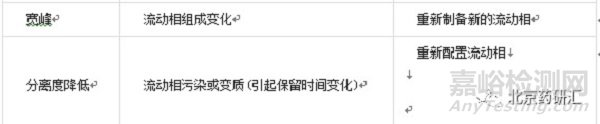
液相色谱系统的很多问题都能在谱图上反映出来,其中一些问题可以通过改变设备参数得到解决,而其它的问题必须通过修改操作程序来解决。
样品溶剂选择不当的解决办法
HPLC溶剂的要求
(1) 高纯度,溶剂与固定相不互溶,并能保持色谱柱的稳定性。由于高效液相灵敏度高,对流动相溶剂的纯度要求也高。不纯的溶剂会引起基线不稳,或产生“伪峰”。
(2) 溶剂对于待测样品,必须具有合适的极性和良好的选择性,即足够的溶解能力。溶剂的极性要与待测样品相匹配,如果溶解度欠佳,样品会在柱头沉淀,不但影响纯化分离,且会使柱子恶化。
(3) 溶剂要与检测器匹配
对于UV-Vis,所用流动相在检测波长下应没有吸收或UV 吸收很小。
(4) 化学稳定性好
不能选与样品发生反应或聚合的溶剂。
(5) 低粘度,沸点适中
减小溶质的传质阻力,降低柱压,利于提高柱效。
常用的低粘度溶剂有丙酮、乙醇、乙腈等;粘度过低的溶剂也不宜采用,如戊烷、乙醚等,易在色谱柱或检测器内形成气泡,影响分离。
HPLC溶剂关键性能
(1) UV吸收性
HPLC级试剂是用特殊方法生产的含有最少的颗粒和最低的紫外吸收物质,除去具有吸收UV的杂质,在规定的波长上吸光度限制在规定值以内。
(2) 梯度洗脱性能
在一个分析周期内程序控制流动相的组成,如溶剂的极性、离子强度和pH值等,用于分析组分数目多、性质差异较大的复杂样品。
采用梯度洗脱可以缩短分析时间,提高分离度,改善峰形,提高检测灵敏度。
① 溶剂的互溶性,不相混溶的溶剂不能作为梯度洗脱的流动相。
当有机溶剂和缓冲液混合时,可能析出盐的晶体,尤其使用磷酸盐时需特别小心。
② 纯度要求更高,以保证良好的重现性。进行样品分析前必须进行空白梯度洗脱,以辨认溶剂杂质峰,因为弱溶剂中的杂质富集在色谱柱头后会被强溶剂洗脱下来。用于梯度洗脱的溶剂需彻底脱气,以防止混合时产生气泡。
③ 防止压力过大。
混合溶剂的粘度常随组成而变化,因而在梯度洗脱时常出现压力的变化。
例如甲醇和水粘度都较小,当二者以相近比例混合时粘度增大很多,此时的柱压大约是甲醇或水为流动相时的两倍。
反向液相色谱

(柱效高、分离能力强、保留机理清楚,是液相色谱分离模式中使用最为广泛的一种。适用于非极性-中等极性组分)
固定相:极性小的烷基键合相 C8柱,C18柱
流动相:流动相极性 > 固定相极性 极性大的甲醇-水或乙腈-水
HPLC里面的溶剂效应是如何产生的,该如何避免?如何判断分叉的峰是否由溶剂效应造成,第一就是通过HPLC仪器比对一下两个劈叉峰的紫外吸收波谱,看看他们是否能完全一样的μV特征吸收,第二将进样体积调节到0.1μl看看,峰是否好转,如果满足上面两个条件几乎就可以肯定是100%的溶剂效应。产生原因:
1、样品溶液的溶剂很可能强于流动相。例如样品溶液的溶剂是100%乙腈(100%的强溶剂),而流动相的组成则较弱,18%的乙腈与72%的水;
2、就是进样量比较大;
3、样品的pH值与HPLC流动相pH悬殊,比如酸性流动相体系,样品pH偏碱性(pH=10),这种情况下也很有可能发生 其实上面三个原因归结起来就是一个原因,那就是样品的溶剂与流动相存在一些不一样,当样品进入流动相的时候,由于这种巨大的差异,一部分样品溶解进入了流动相,一部分还留在进样的溶剂里面,所以在HPLC上出现了保留的差异,所以要解决这个问题的最佳方案就是,使用流动相的起始梯度溶解样品,如果由于溶解度的问题,不得不改用其余的溶剂,那就只能降低进样体积来解决,比如 5μl,1μL。
GC-MS 测定PAHs,HP-5MS (Agilent) 柱,固相萃取后使用甲醇洗脱。用甲醇做溶剂,是否会影响柱效和柱寿命。是否是最佳选择?甲醇的沸点比较高,极性比较大,如果换成正己烷类的弱极性,低沸点,配合一定的色谱条件可以实现溶剂聚焦作用,显著提高保留时间较短的化合物的灵敏度和检出线。甲醇相对于正己烷等极性小的溶剂,造成的柱流失大,国外用乙腈做GC-MS溶剂的文献很多,很多用来减少前面溶剂转换步骤,结合大体积进样等,溶剂先挥发分流走掉,从而进入柱子乙腈溶剂的较少,可认为对柱子影响不大,同时甲醇乙腈对比,乙腈的柱流失要少。
现有个样品,其结构中只有孤立的C=O。由于样品溶于乙醇,所以第一次做紫外扫描时就以乙醇作为溶剂。结果在202nm处测得了最大吸收。检测该化合物的HPLC条件是以乙腈-水作为流动相的。我用流动相作为溶剂再对该化合物进行紫外扫描。结果在197nm处测得了最大吸收。而在200~400nm范围内仅在285nm处测得一很小的吸收峰。在做紫外扫描时,样品溶剂不同,测得的样品的最大吸收波长也会不同。如果用HPLC-UV检测该化合物,波长选为202nm(以乙醇作为溶剂测得的),是否合适呢?在做HPLC检测时,样品的溶剂如果不是流动相之一,会不会影响检测结果呢?C=O在270~300nm有较弱的紫外吸收(ε=10~100),285nm的峰应为C=O的吸收。乙腈-水的极性强于乙醇,由于N-π跃迁的存在,将发生蓝移(最大吸收202变为197),在紫外区的边缘测定有关物质则干扰甚大,不合适;测定含量勉强可以,如果仅测定含量可以根据吸收情况适当增加到210nm左右。如果溶剂跟流动相一样,那紫外吸收也应该一致,否则样品就非完全同一物质。紫外测定选择最大吸收波长进行含量测定,可以提高检测灵敏度,减少其他干扰,但这是相对的。

来源:Internet


