您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2019-10-30 16:03
作者:陈家润 , 江映珠 , 孟兰贞 , 王晓英 , 吴雪龙 , 张一凡
广东省药品监督管理局审评认证中心, 广州 510080
摘要:目的:分析广东省医疗机构中药制剂注册申报存在的问题,为促进医疗机构中药制剂的研发与发展提供参考。方法:通过对2016-2018年60份广东省医疗机构中药制剂注册申报资料进行梳理、归纳,从技术角度阐述医疗机构中药制剂注册申报资料存在的不足。结果与结论:经过技术审评,符合《医疗机构制剂注册管理办法》(试行)有关规定的申报资料24份,通过率为40%。对未通过技术审评的36份注册申报资料的各项目进行系统分析发现,自《医疗机构制剂注册管理办法》实施以来,医疗机构中药制剂研发水平虽有较大程度的提高,但在协定处方使用历史、配制工艺研究、药用辅料使用、直接接触制剂的包装材料和容器选择、申报资料完整性等方面仍存在诸多问题,有较大的提升空间。建议医疗机构引入制剂质量源于设计(quality-by-design,QbD)的研发理念,加大对医疗机构制剂的重视程度;医疗机构制剂注册配套的法规要及时出台,以促进其逐步完善。
关键词:医疗机构制剂 注册 问题 对策
医疗机构制剂(以下简称医院制剂)是指医疗机构根据本单位临床需要经批准而配制、自用的固定处方制剂[1]。医院制剂作为药品的一种特殊补充形式,成为临床用药的有益补充,长期以来,因其便捷、有效等特点在临床诊疗服务中发挥着重要作用。我国制定的《药品管理法》[2]、《医疗机构制剂注册管理办法》(试行)[1]、《医疗机构制剂配制质量管理规范》[3]、《医疗机构制剂配制监督管理办法》[4]、《关于印发加强医疗机构中药制剂管理意见的通知》[5]及其配套技术规范等形成一系列法规体系,有效地规范了医院制剂的注册、配制管理和使用,使医院制剂质量得到相应的保障。医院制剂一般分为中药制剂和化学药制剂,其中以中药制剂居多。根据广东省情况分析,化学药制剂呈逐渐萎缩趋势,而中药制剂受《中医药法》[6]、《关于对医疗机构应用传统工艺配制中药制剂实施备案管理的公告》[7]等政策鼓舞,有快速发展的趋势。由于制剂研发人才短缺、生产设备和检测设备陈旧落后等原因,医院制剂的研发、注册申报存在诸多不完善的地方。本文对2016-2018年广东省医疗机构中药制剂注册申报情况、注册申报资料存在的问题及原因进行分析,为促进中药制剂的开发与发展提供参考。
1 注册申报基本情况
2016-2018年,广东省医疗机构中药制剂注册申报数量为60个,其中有56个品种为协定处方,在申报单位具有至少5年的使用历史而免报临床直接申请配制。经技术审评(包括专家审评),无申报资料不齐全、使用未获批准包装材料、辅料使用不安全、安全及有效性历史佐证缺失、配制工艺研究不充分等重大缺陷,符合《医疗机构制剂注册管理办法》(试行)[1]有关规定的24个,通过率为40%。申报品种的治疗领域为骨科、皮肤科、内科、产科和儿科等;申报单位主要分布在珠三角地区,以广州为主。
2 注册申报资料存在的问题
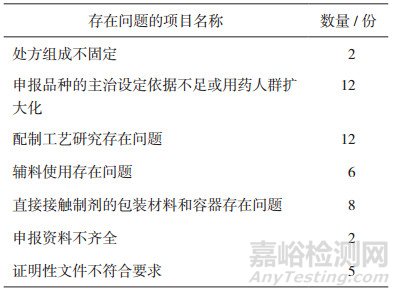
本文对未通过技术审评的36份注册申报资料的各项目进行系统分析,发现协定处方使用历史、配制工艺研究、辅料使用、直接接触制剂的包装材料和容器使用、申报资料完整性等项目存在问题。其中,以申报品种的主治设定依据不足或用药人群扩大化和配制工艺研究两项存在问题最多,各有12份,详见表 1。只存在一个问题的资料26份,存在两个问题的资料8份,存在两个以上问题的资料2份。
表 1 注册申报资料存在问题的项目情况
2.1 协定处方使用历史存在问题
《医疗机构制剂注册管理办法》(试行)[1]规定:根据中医药理论组方,利用传统工艺配制,且该处方在本医疗机构具有5年以上(含5年)使用历史的中药制剂,可免报药效学、毒理学和临床资料,即免报临床直接申请配制。此外,根据《关于印发加强医疗机构中药制剂管理意见的通知》[5]的细化规定:本医疗机构具有5年以上(含5年)使用历史是指能够提供在本医疗机构连续使用5年以上的文字证明资料(如医师处方、科研课题记录、临床调剂记录等),并提供100例以上相对完整的临床病历。提供处方5年使用历史的证明资料的目的是为所申报的医院制剂提供在临床上有效性、安全性的佐证。该项不足表现为以下两种情况:
2.1.1 处方组成不固定
根据广东省医院制剂注册审评标准要求,协定处方组成应基本固定,君臣等主药用量固定,佐使药可根据患者实际情况,用量上允许细微调整。但是,从申报资料看,部分品种的君臣药味种类和用量都存在变化,即该处方治疗某一病症的成熟度不够,无法支撑申报品种的有效性和安全性。
2.1.2 申报品种的主治设定依据不足或用药人群扩大化
参考《药品注册管理办法》[8]对多个适应症中药的每个适应症临床病例数规定,并结合《医疗机构制剂注册管理办法》(试行)[1]对医院制剂临床研究受试例数的规定,广东省在医院制剂注册审评中要求:中药制剂一个主治病症的设定,原则上应至少有60例相关临床疗效证据的支持。但是部分申报品种设定的主治范围过大,找不到符合例数要求的临床病历给予支持。例如,某洗剂申报时的主治范围为用于湿疹、癣、疥疮及虫咬皮疹,但是该处方的临床使用背景并无“疥疮”的主治历史;又如用于治疗儿童感冒退热的某颗粒剂申报时用药范围涵盖1~12岁,但其临床使用历史仅覆盖了6~12岁年龄段的患儿。
2.2 配制工艺研究存在问题
中药制剂配制工艺研究包括提取工艺研究,分离、纯化、浓缩、干燥工艺研究,制剂成型性研究和中试研究等内容[9]。该项不足存在以下三种情况:
2.2.1 提取工艺参数筛选评价指标不足
××颗粒剂提取工艺,针对溶媒种类、提取时间、提取溶媒用量及浓度等工艺参数进行筛选研究,虽然也采用单因素或多因素、多水平等方法,但评价指标仅有出膏率,缺少君、臣等药主要成分保留率等定量指标。
2.2.2 工艺路线不合理
××茶剂,工艺为“部分药材粉碎成粗粉备用,其他药材水提两次,合并煎液,滤过,滤液浓缩,喷雾干燥成干膏粉,再将干膏粉用适量水溶解,均匀喷洒在备用的药材粗粉上,……”,上述干燥成干膏粉再溶解工艺步骤多此一举,不如将其省略,直接将滤液浓缩成固定范围相对密度的清膏后喷洒在药材粗粉上。
2.2.3 小试处方研究结果与中试处方不一致
××贴膏剂,小试处方研究时,粘合剂为聚乙二醇(PEG),在没有任何研究资料支持下,中试处方将粘合剂更换为聚环氧乙烷(PEO)。
2.3 辅料使用存在问题
辅料选择的一般原则:满足制剂成型、稳定、作用特点的要求,不与药物发生不良相互作用,尽量少用或不用[10]。该项不足存在以下两个方面:
2.3.1 辅料种类选择不合理
2.3.1.1 矫味剂与剂型不匹配
某合剂,配制工艺为水煎、浓缩、离心、加矫味剂和抑菌剂、煮沸30分钟、定容、灌封等,矫味剂选用三氯蔗糖。但是,申报单位未考虑三氯蔗糖在高温水溶液条件下会水解产生4-氮-4-脱氧半乳糖及1, 6-二氯-1, 6-二脱氧果糖[11],未对三氯蔗糖经高温煮沸后的水解情况分析及其水解产物的安全性进行评价。
2.3.1.2 抑菌剂未考虑适用范围
某合剂,质量标准拟定pH范围为4.0-6.0,3批样品实测pH 4.6-4.8。选用抑菌剂为0.3%的苯甲酸钠,但是中药制剂常识[9]提示:苯甲酸钠一般在pH 4以下时防腐作用较好,pH超过5时,用量不得少于0.5%。本品拟定的pH 4.0-6.0范围,当pH超过5时,在现有抑菌剂浓度下,是否依然发挥抑菌效果,缺乏足够的支持数据。
2.3.2 辅料用量不符合规定
依据《中华人民共和国药典》(以下简称《中国药典》)[12]制剂通则颗粒剂项下规定:除另有规定外,中药饮片应按各品种项下规定的方法进行提取、纯化、浓缩成规定的清膏,采用适宜的方法干燥并制成细粉,加适量辅料(不超过干膏量的2倍)或饮片细粉,混匀并制成颗粒,即辅料用量不超过干膏量的2倍。从安全性角度考虑,辅料用量不得超过美国食品药品监督管理局(Food and Drug Administration,FDA)、国家药品监督管理局药品审评中心(Center for Drug Evaluation,NMPA,CDE)推荐用量。
2.3.2.1 颗粒剂辅料用量超干膏量2倍
某颗粒剂,填充剂为蔗糖(同时为矫味剂)与糊精,二者的用量与浸膏粉比例为“辅料:浸膏粉=4.2:1”,不符合《中国药典》四部制剂通则颗粒剂的规定“辅料加入量不超过干膏量的2倍”。
2.3.2.2 辅料用量超过FDA、CDE推荐用量
某化痰颗粒剂,填充剂为蔗糖(同时为矫味剂)与倍他环糊精,申报品种中试样品中的倍他环糊精的日服用量为32.5~41.6 g/day。笔者以“倍他环糊精”分别检索CDE药用辅料数据库[13]和FDA非活性成份数据库[14],检索结果显示倍他环糊精的口服最大日用量分别为30 mg/day和133.3 mg/day。申报品种倍他环糊精的日服用量均显著高于以上标准,申报单位应对该申报品种辅料用量的安全性进行合理评价。
2.4 直接接触制剂的包装材料和容器存在问题
根据《医疗机构制剂注册管理办法》(试行)规定:医疗机构制剂注册申报材料要求,申报资料项目3证明性文件要求提供“直接接触制剂的包装材料和容器的注册证书复印件”,且必须为批准文号在有效期内的注册批件。但是,根据《总局关于药包材药用辅料与药品关联审评审批有关事项的公告》(2016年第134号)(以下简称“第134号公告”)[15]规定,药包材由单独审评改为在国家局审评药品注册申请时一并审评审批,即药包材不再单独核发相关注册批准证明文件(包括新注册批准文件和再注册批准文件),由此造成医院制剂注册审评过程中出现以下问题:
2.4.1 药包材再注册批准文件已过有效期
某合剂,直接接触制剂的包装材料和容器为口服液体药用聚酯瓶,申报单位提供的口服液体药用聚酯瓶包材注册证已过期,且经查询CDE原料药、药用辅料和药包材登记信息公示也无该药包材的关联审评信息。
2.4.2 使用未获得批准文号的药包材
某贴膏剂,该制剂背衬材料为无直接接触制剂的包装材料和容器的注册证书的无纺布,该包材的执行标准为非织造布行业标准。
2.5 其他问题
2.5.1 申报资料不齐全
根据《医疗机构制剂注册管理办法》(试行)规定,符合免报临床直接申请配制条件的品种,如果处方组成含有法定标准中标识有毒性及现代毒理学证明有毒性的药材,仍需报送毒理学资料(急性毒性试验资料和长期毒性试验资料)。审评过程中发现有品种处方中含有药典标注有小毒的苦杏仁,并没有报送毒理学研究资料。
2.5.2 证明性文件不符合要求
根据《医疗机构制剂注册管理办法》(试行)规定:未取得《医疗机构制剂许可证》或《医疗机构制剂许可证》无相应制剂剂型的“医院”类别的医疗机构申请医疗机构中药制剂,还应当提供委托配制中药制剂双方签订的委托配制合同。但是调查发现,某合剂,申报单位委托某中医院进行样品的试制,并提供了《委托配制协议书》复印件,协议书中无样品检验、质量保证等相关方面的协议内容;据进一步了解,受托方某中医院并不具备微生物限度检测能力,不能对样品进行全检。
3 原因分析
3.1 医疗机构制剂研发人员理念陈旧
从医疗机构制剂注册资料及与从事医疗机构制剂研发人员的交流中获知:有相当部分的医疗机构制剂研发人员对制剂研发的科学性、严谨性认识不足,认为医疗机构制剂能配制出来且检验合格就行,不属于研发新药,其研究的科学性、规范性不需要严格执行。这是典型的制剂质量是检验出来(或质量源于检验,quality-by-testing,QbT)的理念,与现在国际流行的制剂质量是设计出来(或质量源于设计,quality-by-design,QbD)的理念相违背。殊不知制剂研发在处方、工艺设计环节就存在安全性缺陷,生产与检验阶段是无法弥补的。
3.2 医疗机构对医院制剂研发的重视程度不足
由于受各种因素制约,医疗机构制剂的售价一直较低,再加上批量较小、有效期较短,医院制剂的综合生产成本较高、利润较微薄。因此,制剂部门在医疗机构中的地位一般较低,较难配备充足的高素质研发人员与获得足够发展资金的支持,而没有足够资金的支持与投入,就很难进行研发场地的改造、研发设备的更新换代、研发人员的招聘与培训教育提高。
3.3 医疗机构制剂注册配套的法规滞后
原国家食品药品监督管理总局发布第134号公告以来,药包材由单独审评改为在国家药品监督管理部门审评药品注册申请时一并审评审批,药包材不再单独核发相关注册批准证明文件(包括新注册批准文件和再注册批准文件)。但是,对于再注册证已经过期或未获得注册证的药包材要在医疗机构新制剂中使用的情况该如何处理,国家药品监督管理部门没有相应的配套处理方案,省药品监督管理部门也不知道该如何做,毕竟根据第134号公告药包材关联审评的权限在国家层面,省级层面无权限进行药包材关联审评。
4 对策与建议
4.1 引入QbD的制剂研发理念
美国FDA于2001年在药品管理中率先引入QbD质量管理理念,ICH在Q8中也强调药物研发过程中要贯彻QbD理念。QbT与QbD理念的区别[16]:在传统的QbT模式下,制剂的质量主要通过终产品的检验等方式实现,偏重于下游控制,类似水灾治理中的“堵”法;而在QbD理念中,则强调质量的风险管理,将质量控制提前至研发阶段,类似水灾治理中的“疏”法,较传统的QbT系统更为主动、有效。因此,为在源头上保证医疗机构制剂的安全性和有效性,杜绝辅料使用等方面存在的设计缺陷,建议在医疗机构制剂研发阶段引入QbD质量理念。
4.2 加大对医疗机构制剂的重视程度
现阶段,药品在医院销售取消“零加成”、《中医药法》[6]出台以及传统中药制剂实行备案制的大环境下,医疗机构中药制剂发展大有可为。因此,医疗机构要改变以往“医院制剂生产使用就是赔钱的买卖”的观念,加大对医院制剂关注力度,重视医院制剂的开发。在日常临床过程中,医生应注意收集计划开发协定处方的处方、病历、使用疗效和不良反应等信息,并将上述资料定期移交医院注册申报部门进行汇总、分析、归档,为注册申报打下坚实的临床基础。基于上述前期准备工作,戴春光等[17]认为注册申报部门可适时启动制剂开发立项可行性评估工作。对于通过可行性评估的协定处方,可以通过委托有资质、有能力的药物研发机构研究开发或筹建自己的研发团队等形式,将协定处方开发成医院制剂,若医院制剂疗效和安全性都较好,可以进一步开发成中药新药。
4.3 医疗机构制剂注册配套的法规要及时出台
针对再注册证已经过期或未获得注册证的药包材要在医疗机构新制剂中使用情况的处理,建议国家药品监督管理部门出台相关政策明确处理措施;针对没有专门的医疗机构制剂研究技术指导原则来指导研究情况的,建议国家层面出台符合医疗机构制剂特点、操作性强的研究技术指导原则,以指导医疗机构制剂的研究、统一全国医疗机构制剂的审评审批尺度。
参考文献
[1] 国家食品药品监督管理局.国家食品药品监督管理局令[2005] 20号医疗机构制剂注册管理办法(试行)[S]. 2005.
[2] 全国人民代表大会常务委员会.中国人民共和国药品管理法[S]. 2015.
[3] 国家药品监督管理局.国家药品监督管理局令[2001] 27号医疗机构制剂配制质量管理规范[S]. 2001.
[4] 国家食品药品监督管理局.国家食品药品监督管理局令[2005] 18号医疗机构制剂配制监督管理办法[S]. 2005.
[5] 卫生部, 国家中医药管理局, 国家食品药品监督管理局.国中医药医政发[2010] 39号关于印发加强医疗机构中药制剂管理意见的通知[S]. 2010.
[6] 全国人民代表大会常务委员会.中国人民共和国中医药法[S]. 2016.
[7] 国家食品药品监督管理总局. 2018年第19号关于对医疗机构应用传统工艺配制中药制剂实施备案管理的公告[S]. 2018.
[8] 国家食品药品监督管理局.国家食品药品监督管理局令[2007] 28号药品注册管理办法[S]. 2007.
[9] 张兆旺.中药药剂学新世纪(第二版)[M].北京: 中国中医药出版社, 2017: 549;48-49.
[10] 北京市食品药品监督管理局.京食药监药注[2014] 24号北京市食品药品监督管理局关于印发北京市医疗机构化学制剂配制工艺研究等9个技术指导原则的通知[S]. 2014.
[11] R.C.罗, P.J.舍斯基, P.J.韦勒.药用辅料手册[M].北京: 化学工业出版社, 2005: 711-712.
[12] 中华人民共和国药典: 四部[S]. 2015: 7.
[13] 国家药品监督管理局药品审评中心.辅料数据库[DB/OL].(2012-03-06)[2019-05-10]. http://www.cde.org.cn/drugInfo.do?method=fuliaoView&flId=257.
[14] 美国食品药品监督管理局. FDA批准药物非活性成分数据库[DB/OL].[2019-05-10]. https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm?event=BasicSearch.page.
[15] 国家食品药品监督管理总局. 2016年第134号关于药包材药用辅料与药品关联审评审批有关事项的公告[S]. 2016.
[16] 王明娟, 胡晓茹, 戴忠, 等. 新型的药品质量管理理念"质量源于设计"[J]. 中国新药杂志, 2014, 23(8): 948-954.
[17] 戴春光. 医院制剂申报注册的体会[J]. 中医药导报, 2017, 23(18): 61-62.

来源:中国药事


