您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-06-27 12:40
1.路径1→MHLW approval(承認)
适用器械
新分析物
有approval standards的class III
没有标准的class III
不符合对应标准的class I / II / III
注册方式
PMDA审核提交资料,MHLW最后批准
提交内容
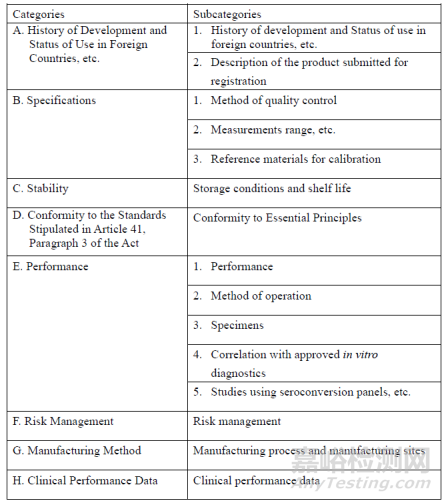
申请表和技术文档,技术文档内容见下表:
周期
需要专家审核,约12个月
常规产品,约7个月
费用
454,800-2,534,000日元(伴随诊断4,295,000日元)
2.路径2→RCB certification(認証)
适用器械
有认证标准并符合的Class II
注册方式
第三方认证机构Registered Certification Body (RCB)进行认证,认证完成会在网上公布。
RCB的最新清单可查询:
www.mhlw.go.jp/stf/seisakunitsuite/bunya/kenkou_iryou/iyakuhin/touroku/index.html
认证流程
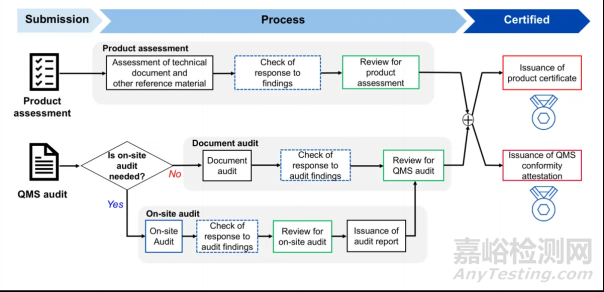
需要先向RCB提出认证申请,然后进行产品评审和QMS审核。流程这里借用TÜV南德对外公布的认证流程。
3.路径3→PMDA notification(届出)
适用器械
符合对应标准的Class I
注册方式
线上向PMDA提交资料,不会审核
提交内容
器械分类、名称、预期用途、外形结构和原理、主要反应成分、产品规格、使用方法,存储方法及有效期、制造方法、制造设施。
周期
提交当日
费用
免费
临床性能研究
原则上,除非申请的是新产品,否则不需要临床性能数据。
应包括至少2个地点,150个样本(包括正常范围内的样本)。少于150个的标本数量是可以接受的,如果这个数量可以保证统计分析和足够的临床评价。
原则上可以使用在日本以外进行的临床性能研究的结果。然而,应考虑种族差异、环境因素和临床实践差异等因素对拟议IVD的性能和临床意义的影响,决定是否使用在日本以外进行的研究结果。

' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
来源:Internet


