一、我国法律法规体系框架
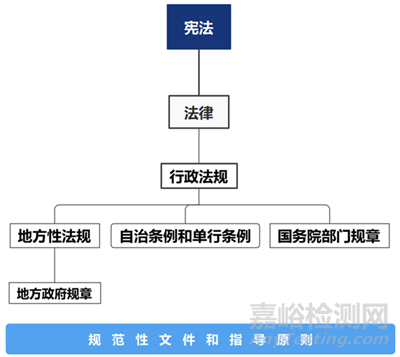
我国的法律法规体系以宪法为统帅,以法律为主干,以行政法规和地方性法规为重要组成部分,具体分为7种立法形式:
1)宪法:最高法律层级的最高法,由全国人民代表大会制定。
2)法律:由国家制定或认可并依靠国家强制力保证实施的,反映由特定社会物质生活条件所决定的统治阶级意志,以权利和义务为内容,以确认、保护和发展对统治阶级有利的社会关系和社会秩序为目的的行为规范体系。
3)行政法规:由国务院审议通过并颁布,一般以实施条例作为结尾,法律效力低于宪法和法律。
4)地方性法规:省、直辖市人民代表大会制定并颁布,在本辖区内有效,效力低于法律和行政法规。
5)自治条例和单行条例:自治区的,由自治区人民代表大会制定,报全国人大(常委会)批准生效;自治州、自治县的由当地人民代表大会制定,报省或自治区人大批准生效,报全国人大常委会备案。在本辖区内有效,法律效力低于法律和行政法规。
6)国务院部门规章:由国务院的部委、直属机构等制定并发布实施,多以管理办法为结尾,法律效力低于法律和行政法规。——在药监体系中,由国家市场监督管理总局制定发布,如《药品注册管理办法》、《药品生产监督管理办法》。
7)地方政府规章:地方政府制定,在本辖区内有效,法律效力低于统计或上级的地方性法规。
此外,再往下还有众多规范性文件和指导原则进行补充和完善,以指导具体实施。——具体到药监体系,包括NMPA颁布的2016年134号文、2017年146号文,以及2019年56号文;以及CDE颁布的《化学原料药、药用辅料及药包材与药品制剂关联审评审批管理规定(征求意见稿)》。
从法律效力层级分开,可以归纳为下图:
二、我国原辅包药监政策静态分布
将原辅包相关法律法规等制度文件放入到我国的法律法规体系之中,则形成以下俯视分布图:
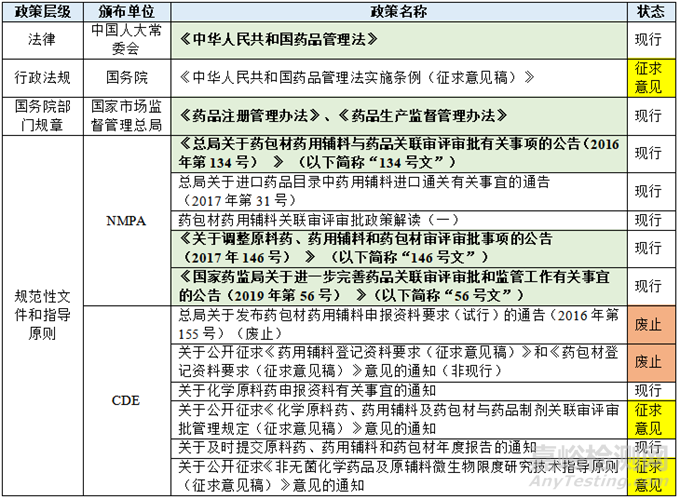
其中用浅绿色背景标注的制度文件为目前指导原辅包关联审评审批制度实践的核心制度,我会在下文予以解读。
若从时间脉络,去动态地梳理相关法律法规的历史变迁,则可以用这张图来体现:
四、核心政策解读
4.1《中华人民共和国药品管理法》
《中华人民共和国药品管理法》中涉及原辅包的条款主要包括3条,分布为第二十五条、第四十五条和第四十六条。相应条款及解读如下:
解读:
1)虽没有关联审评审批的明确字眼,但实质内容已明确:原料药在制剂审批时,一并进行审评审批;药用辅料和药包材在制剂审批时,一并进行审评,无需审批。
2)明确辅料的定义,即生产药品和调配处方时所用的赋形剂和附加剂。
解读:
1)明确生产药品的原辅包应符合药用要求。何为药用要求呢?在此处并没有明确规定,但是在56号文中有明确:药用要求即辅料的质量、安全及功效需满足药品的要求。
2)明确生产药品的原料药和辅料,要符合GMP要求,但对药包材却没有此要求。原料药需满足GMP要求大家可能比较了解,但对于药用辅料也要满足GMP要求可能会比较陌生,更何况药用辅料有一个《药用辅料生产质量管理规范》(2006版)。基于这一条的要求,药用辅料企业需按照GMP要求对药用辅料生产、质量进行管理。
4.2《药品注册管理办法》
《药品注册管理办法》以规范药品注册行为,保证药品的安全、有效和质量可控为目的;针对在我国境内以药品上市为目的,从事药品研制、 注册及监督管理活动而制定的部门规章。其不仅在第十四条中明确使用了“关联审评审批制度”这个描述,还在第三章第三节对关联审评审批进行了详细规定。
解读:
明确NMPA对原辅包建立关联审评审批制度进行管理,同时明确CDE建立原辅包信息登记平台,用于原辅包登记人进行信息登记和公示,制剂申请人或MAH选择原辅包。
解读:
明确了原辅包申请人和CDE的职责,前者负责完成研究,并进行登记申报;后者负责公示信息,以及关联审评。
解读:
明确了药品制剂申请人的两种选择路径:选择已登记的原辅包和选择未登记的原辅包。针对前者,无需提供研究资料;针对后者,需一并提交研究资料。
解读:
1)明确关联审评过程中,针对补充资料的提交要求,即按照补充资料程序执行;
2)明确会基于风险对原辅包企业进行延伸检查,保留了核查的权利。虽然在实际执行中进行延伸检查的概率不大,但是若做得太差,导致风险偏高,也是有可能的,所以原辅包企业还是要尽可能将研究做充分;
3)提出了一个可以单独审评审批的情况,即仿制境内已上市药品所用的化学原料一年,可申请单独审评审批。
解读:
对审评审批结果进行了规定,具体归纳如下:
|
|
|
通过
|
原料药:登记状态由“I”状态变为“A”状态;颁发化学原料药批准通知书(含登记号信息)、核准后的生产工艺、质量标准和标签。
药用辅料和药包材:登记状态由“I”状态变为“A”状态。
制剂:批准通知书、核准后的生产工艺、质量标准和标签。
|
|
不通过
|
原料药:登记状态保持“I”状态不变;不予批准,并颁发化学原料药不予批准通知书。
药用辅料和药包材:登记状态保持“I”状态不变。
制剂:不予批准,并颁发不予批准通知书。
|
|
原料药单独审评审批
|
|
通过
|
颁发化学原料药批准通知书(含登记号信息)、核准后的生产工艺、质量标准和标签。
|
|
不通过
|
颁发不予批准通知书
|
4.3《药品生产监督管理办法》
《药品生产监督管理办法》以加强药品生产监督管理,规范药品生产活动为目的,针对在我国境内上市药品的生产及监督管理活动而制定的部门章程。涉及原辅包管理的条款包括:
解读:明确监管机构为省局级别;监管对象包括原辅包供应商、生产企业;监管类型包括日程监督检查和必要的延伸检查。

解读:明确监管频率:每年抽检,五年内必检一次。
解读:违法处方要求如下:
|
处罚级别
|
处罚内容
|
|
第一档
|
限期改正,给予警告。
|
|
第二档
|
逾期不改,处10~50万罚款
|
|
第三档
|
情节严重,对企业:
● 处以50~200万罚款,
● 责令停产停业整顿直至吊销药品批准证明文件、药品生产许可证、药品经营许可证,对于药物非临床安评机构,临床试验机构,五年内不得开展相关研究或试验;
对法定代表、主要负责人、直接负责的主管人员和其他责任人员:
● 没收违法期间的收入
● 处所获收入10%~50%的罚款
● 10年或终身禁止从事药品生产经营活动。
|
4.4 134号文&156号文&56号文
NMPA发布的《总局关于药包材药用辅料与药品关联审评审批有关事项的公告(2016年第134号) 》 、《关于调整原料药、药用辅料和药包材审评审批事项的公告(2017年146号) 》 和《国家药监局关于进一步完善药品关联审评审批和监管工作有关事宜的公告(2019年第56号) 》是目前指导原辅包关联审评审批具体实施最核心的规范性文件,三者内容上有重合、有替代、有交叉,共同指导原辅包关联审评审批。














