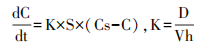摘要
粒度和粒度分布是原辅料以及药物颗粒的关键质量属性,对药物制剂的生产可行性和产品性能有着重要的影响。本文旨在通过对口服固体制剂粒度和粒度分布与生产可行性、产品性能之间的相关性进行剖析,提出粒度和粒度分布分析方法开发和验证过程的关键考虑因素,并探讨如何建立合理的粒度分布控制标准,为药物研发和生产阶段进行粒度和粒度分布研究提供参考。
在口服固体制剂的制备过程中,药物粉体的粒度分布(particle size distribution,PSD)是产品开发和质量控制的关键参数之一,几乎影响整个生产过程。粉体的粒度对于产品的溶出度、生物利用度以及稳定性等性能具有显著影响。例如,减小难溶性药物颗粒粒度时,比表面积增加,使得溶剂更易包围颗粒,显著提高溶出速率[1]。此外,原辅料的粒度及粒度分布对于物料的流动性、可压性、混合均匀性、剂量单位均匀性等特性也会造成影响。例如,微粉化后的原料药颗粒之间静电作用较强,流动性较差,可能出现颗粒结团现象,造成原辅料混合不均匀[2]。药物粉体的粒度分布对于产品的质量和有效性均有深远的影响,应在药物研发及生产过程的整个生命周期中予以关注,并建立有效的控制措施。
目前,药品质量管理的理念已经从“药品质量是通过检验来控制”到“药品质量是通过生产过程控制来实现”,进入到“药品质量是通过良好的设计而生产出来”[即“质量源于设计”(quality by design,QbD)]的阶段[3]。这也就意味着药品从研发开始就要考虑不同因素对最终产品质量的影响。QbD理念的引入提示药品生产企业应该基于药物的处方和生产工艺,评估原料药、辅料以及药物粉体的粒度分布对产品关键质量属性(critical quality attribute,CQA)的影响。如果在药物生产的不同工序阶段(例如,过筛、混合、制粒、压片、包衣等工序),某些原料药或药物粉体被认为是对CQA有影响的关键物料属性(critical material attribute,CMA),则有必要对这些物料的粒度分布进行研究,并根据风险评估结果建立必要的控制措施。此外,开发合理的粒度分布分析方法是进行粒度分布研究的基础。QbD的策略不仅可以应用于药品生产全过程,也可拓展到分析方法的开发和验证过程中进行应用[4]。粒度分布研究方法不同于有关物质检测等方法,其方法重现性受取样、分散介质等多种因素的影响。基于QbD理念进行粒度分布分析方法的设计和确认,可保证分析方法的科学性、准确性和可行性,通过分析方法的持续确证,始终如一提供准确的分析数据,有效控制药品质量。
本文旨在探讨口服固体制剂药物粉末的粒度分布与生产可行性、产品性能之间的相关性,重点关注开发粒度分布分析方法和验证过程的关键考虑因素,以及如何建立合理的粒度分布控制标准的实践应用。
一、粒度分布与生产可行性、产品性能的相关性
粒度分布是制备过程中非常重要的质量指标,对于药物制剂的生产可行性和产品性能有着显著的影响。首先,粒度分布的合理控制可以提高药品的生产可行性。在制药过程中,颗粒大小不同会影响材料的流动性、混合均匀度以及成型压力等质量指标或生产参数,从而进一步影响产品的生产可行性。其次,粒度分布与产品性能密切相关。颗粒较小会导致表面积增大,从而使得药品溶解度更高,吸收更快且更彻底,但同时也可能引发颗粒聚集、崩解不良等问题;颗粒较大则有助于减少颗粒聚集、保持稳定性,但可能导致疏松性差、难以混合等问题。因此,只有在对粒度分布进行全面有效地控制后,才能生产出合格的高质量药品。应该根据所生产的药品类型和特点,结合现有的制造工艺和设备,合理分析粒度分布与生产可行性、产品性能的相关性,以此帮助评估何时需要对粒度分布进行研究,并采取措施确保工艺稳定和产品质量可控。
1.1 原辅料药性质
原料药的粒度是制剂开发中的关键物料属性,对药物的粉体学特性、制剂的均匀性、原料药和制剂的化学稳定性以及制剂的溶出性能产生影响,进而影响制剂在人体内的释放和吸收,最终影响制剂的生物利用度和药物疗效。
药物溶出的规律通常可通过Noeys-Whitney方程式来描述。影响溶出速率的主要因素包括颗粒的表面积、药物的溶解度和溶出速率常数,其中溶出速率常数与扩散系数、介质体积和扩散层厚度相关。对于属于生物药剂学分类系统(biopharmaceutical classification system,BCS)2/4的低溶解性药物,常常采用微粉化工艺对原料药进行预处理,以减小颗粒大小,增大表面积S,从而提高药物的溶解速率,最终提高制剂的溶出速率和生物利用度。
K:溶出速度常数;D:药物扩散系数;h:扩散边界厚度;V:溶出介质的量;S:溶质的表面积;Cs:药物的溶解度;C:溶液主体中药物浓度,若满足漏槽条件,则 C=0。
随着药物颗粒尺寸的减小,相应地会引发粉体学相关问题。颗粒尺寸的减小会增加粉体之间的分子引力和静电作用力,使得颗粒更容易聚集成团,增加黏结性,同时休止角也增大,从而导致粉体的流动性下降。
原料药的粒度和粒度分布也会影响制剂的含量均匀性。特别是对于小规格制剂而言,其粉体混合不均匀的风险更高。已有研究证明[5],原料药的粒度是对生产工艺稳定性影响最大的关键质量属性。Rochers等[6]建立了预测模型,用于预估满足USP<905>含量均匀度要求时,颗粒粒度及粒度分布应达到的控制范围,证明了原料药的粒度和粒度分布与制剂的含量均匀性之间存在函数关系。
辅料作为制剂的重要组成部分,也从诸多方面影响着产品质量。尤其对于缓控释制剂中的关键辅料,其粒度、微观形态等可能影响粉体的流动性、可压性,甚至影响缓释制剂的释放行为。羟丙甲纤维素(hypromellose,HPMC)常作为缓控释制剂的功能性辅料。有研究表明[7],HPMC颗粒大小的调整可能存在“阈值效应”,对某些制剂而言,当HPMC颗粒大小超过关键阈值时,其影响表现为更快的体外药物释放速度和明显变化的药物释放机制。
1.2 制剂工艺
口服固体制剂常用的生产工艺有粉末直压、干法制粒和湿法制粒。药品生产企业应基于原料药和辅料的性质进行风险评估,选择合适的生产工艺。
粉末直压工艺只涉及物料混合和直接压片两个步骤,生产过程简单、节能省时,同时降低工艺条件对产品质量的风险,但对原料药和辅料的相关性质要求更高。这些性质不仅仅要满足工艺的生产需求,还需要在生产过程中保持一致。为确保粉体具有适当的粒度分布、密度、流动性以及可压性,粒度、粒度分布等控制显得更加重要。例如,在混合不同组分时,如果颗粒的大小差异显著,可能导致混合物料分层现象。即使最终混合物的均匀度符合标准,但在后续的转运、储存或压片过程中,仍可能发生物料分层或结团现象。为保证生产过程中物料状态的一致性,有必要对关键物料制定粒度控制标准。
制粒工艺本质上是将分散的粒子制备成更大颗粒的过程,原料药与辅料的粒子在一定程度上可以经过“改造”,相较于粉末直压工艺,制粒工艺对原辅料的性质更具有包容性。然而,与此同时也增加了工艺条件对产品质量带来的相关风险。后续的压片过程直接受制粒颗粒的影响,包括颗粒的流动性、可压性,以及与产品性能相关的药物溶出度和生物利用度等,均与颗粒粒度分布、孔隙率等性质相关。
美国食品药品监督管理局(FDA)颁布的《Guide to Inspection of Oral Solid Dosage forms: Pre/Post Approval Issues for Development and Validation》[8]中曾指出,颗粒的粒度分布是判断制粒工艺批间一致性的重要物理参数,尤其对于临床批次和商业生产批次的对比,或是生产工艺变更前后样品的对比,粒度分布数据是证明工艺的稳健性和产品质量可比性的重要依据。《美国药典》(USP)43<711>中规定[9],当药物的颗粒大小分布较为均匀时,可以通过粒度分布来代替溶出度的测定,间接证明了颗粒的粒度分布对产品质量和有效性的影响。
综上,需综合考虑原料药特性、生产工艺与粒度分布的相关性,结合QbD理念进行合理的工艺设计和工艺控制,通过优化处方和生产工艺来改善药物颗粒的粒度分布,提高药品的质量和稳定性。
二、粒度及粒度分布检查的法规要求和指南概述
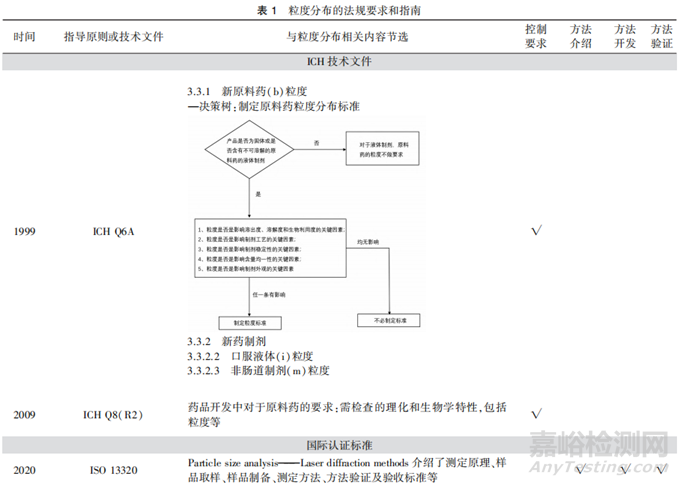
《化学药物制剂基本研究技术指导原则》[10]中对原料药、辅料和药物颗粒的粒度均进行了要求,强调了粒度可能对药物溶解性能、制剂的含量均匀度,或生物利用度及临床疗效产生影响。ICH从QbD角度,在ICH Q6A指南(决策树#3)中提出了何时需对原料药制定粒度标准的建议[11],对于影响溶解度、溶出度、生物利用度、制剂工艺、制剂稳定性、含量、制剂外观等的原料药,应通过适当方法测定粒度及粒度分布,并建立合理的控制限度。在ICH Q8(R2)[3]指南中提出在药品开发的过程中需要检查原料药的粒度等理化及生物学特性,综合考虑这些特性间的关联性。上述指导原则或指南提出了需对粒度进行控制的要求,但未对粒度分析方法及方法学验证的相关内容进行规定。
各国药典、ISO13320等文件提供了粒度分析方法及验证要求的相关内容(见表1)。USP43<786>和<429>章节[9]、《欧洲药典》(EP)11.0 2.9.31章节[12]明确了粒度分布测定分析方法开发和验证等相关内容。其中,USP43<429>和EP 11.0 2.9.31基于ISO13320(1999)和ISO9276-1(1998)开发,国内推出了相应的GB/T 19077粒度分析-激光衍射法。目前,ISO13320已更新至2020年版[13]并引入了QbD理念进行分析方法的开发和验证。《中国药典》2020年版(四部)通则<0982>[14]对粒度分布测定仪器的一般要求和分析方法进行了简单介绍,但未明确分析方法验证等内容。当前国内缺乏粒度分析方法开发和验证的相关指导原则,然而由于粒度分布是药物的关键质量属性之一,药品生产企业应参照国际规定,进行粒度分布研究并提供必要的分析方法学验证资料,以确保粒度分布测定结果的可靠性。
三、粒度分布分析方法开发及验证
粒度分布的测定方法有筛分法、显微镜法、光散射法等,每种方法都有其各自的优缺点,并且适用于不同类型的粒子或颗粒物质。其中,筛分法原理简单、操作方便,常用于测量较大颗粒的粒度分布。然而,由于筛分法的粒度分布划分受限于筛网数目的设置,在一定程度上影响了测量结果的准确度,尤其对于难溶性药物或经评估对产品质量有较大影响的药物颗粒,即使微小的粒度和粒度分布变化也可能对产品质量产生重大影响,需采用更高准确度的方法来测量粒度和粒度分布。
光散射法中的激光衍射法具备测定范围广(0.1~3 000 μm)、快速、自动化等许多优点,特别是在测定极细微颗粒的性能方面表现突出,已经成为目前主流的粒度分布测定方法之一。以下将以激光衍射法为代表,对粒度分布测定方法的开发和验证进行探讨。
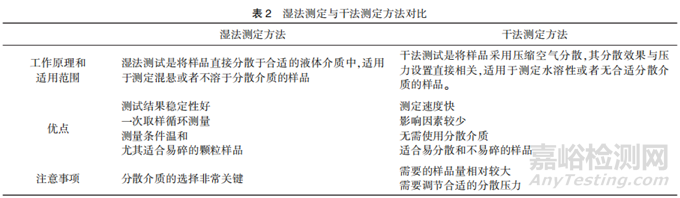
激光衍射法的工作原理是单色光束照射到颗粒后发生衍射现象,由于衍射光的能量分布与颗粒的大小有关,通过测定衍射光的能量分布,根据米氏散射理论和弗朗霍夫近似理论,可计算出颗粒的粒度分布。根据分散介质的不同,激光衍射法可分为湿法测定(分散介质为液体)和干法测定(分散介质为气体)。湿法测定用于测定混悬供试品或不溶于分散介质的供试品,干法测定用于测定水溶性或无合适分散介质的固态供试品[14]。湿法测定的粒度范围宽、测试结果稳定,是更常用的测定方法。但对于容易溶解于分散剂中的颗粒来说,干法通常为唯一选择,且干法测定速度更快。湿法和干法测定的优势对比情况详见表2。
3.1 分析方法开发
3.1.1 取样
对于任何一种颗粒表征技术来说,从较大的样品量中选取具有代表性的样品都是一个挑战。进行激光衍射法检测时,采样问题可能会引起检测结果的较大偏差,特别是在测定大颗粒样品或接近粒度分布边界参数(如D95)时更为明显。这是因为激光衍射法作为一种基于样品体积的测量技术,对所选取样品中粗颗粒数量的微小变化非常敏感。随着颗粒大小和分布宽度的增加,采样对重现性的影响也会增加。因此,为了确保可重现的结果,需要保证足够量的取样量(通常大于1~2 g)[15]。随着颗粒尺寸的增加,达到可重现结果所需的最小样品量也需随之增加。此外,不同的取样方法对检测结果的影响有很大差异,选择合适的取样设备将大大提高粒度测定的重现性[16]。USP43<429>和ISO13320建议采用例如旋转缩分器或锥形四分法等样品分离技术进行代表性样品制备。ISO 14488[17]提供了Spinning Riffler、Static Riffle Divider、Coning and Quartering、Increment Division Method等多种样品分离技术的适用范围、使用建议等详细信息,可供参考研究。
3.1.2 分散体系
建立合适的分散体系对于样品的检测也是至关重要的。样品分散的目的是尽可能地削弱颗粒之间的聚集,尤其对于粒度小或是黏度大等易发生聚集的颗粒,应采用合适的分散介质和分散条件使颗粒能均匀分散,同时也应避免过度使用分散能量造成颗粒的损耗,导致检测结果的偏差。
关于气体中的分散,应使用适当的干式分散器。对于颗粒较大、流动性较好的颗粒,通常通过重力自由下落足够实现分散。对于聚集的颗粒,通常需要利用压缩气体或真空条件,在颗粒之间或颗粒与器壁之间发生碰撞的剪切应力作用下进行分散。
关于液体中的分散,可参考ISO 14887[18]进行液体分散介质的选择。通常可以使用搅拌或超声来促进颗粒在液体中适当分散,可以借助显微镜对分散体系的质量进行初步判定。此外,需关注分散介质体积等因素对测量结果的影响。液体分散介质的体积随着待测颗粒粒度分布的宽度增大而增加[19]。例如,要使D90的精密度RSD达到3%以内,对于0.3 mL体积、粒度范围为2~200 μm范围的待测颗粒,至少需要500 mL悬浮液才能达到目标精密度[13]。粒度分布研究可根据目标精密度来确定适当分散条件,以保证测定结果的重现性。
3.1.3 测量条件
为了获得可重复的粒度测量结果,需要确保样品处于稳定的分散状态。对于湿法分散,能量来源于分散池中的搅拌器、泵和超声波。通过调整搅拌速度、泵速和超声强度等参数,可以有效地实现颗粒的均匀分散,并避免颗粒破碎。此外,还需要根据颗粒的大小和性质确定遮光度范围。湿法测量和干法测量所需要的供试品量通常应分别达到检测器遮光度范围的8%~20%和0.5%~5%[14]。干法分散的能量主要来源为压缩空气流。ISO 13320指出,理想的分散压力可使粒度在一定范围内保持不变,表明颗粒已均匀分散且未被破坏。但是,理想状态较难实现。因此,可将干法分散的结果与充分分散的湿法测量结果进行比较,当两种方法结果接近一致时,说明选取的分散压力满足测定要求。此外,进样速度、测量时间、文丘里管能量等因素也会导致结果的变异。
综上所述,任何颗粒表征系统要获得可重复的结果,需综合考虑取样的代表性、稳定的分散状态以及适当的测量条件。在分析方法开发过程中,应根据QbD理念确定检测目的,结合样品性质对影响因素进行风险评估,并确定关键测量参数。此外,还需要关注分析仪器类型和光学模型等方面的信息。选择不合适的光学模型或折射率值可能会导致粒度分布结果的显著偏差[4]。为了获得更好的重现性,有必要明确光学模型和光学参数等信息。同时,建议将粒度分布分析方法开发过程的研究数据纳入申报材料,并在质量标准的粒度分布项中提供详细的取样、样品分散和测量条件方面的具体操作描述。
3.2 分析方法验证
由于粒度分析方法的独特性,其方法验证内容与ICH Q2A、ICH Q2B和USP43通则<1225>中描述的典型的验证内容有所不同。ICH定义的验证参数如专属性、定量限、检出限、线性、范围等,在粒度分析方法的验证中通常不适用。粒度分析方法的验证通常需要对精密度(重复性、中间精密度)和方法耐用性进行验证。
USP43<429>、EP11.0 2.9.13、《中国药典》2020年版<0982>第三法(光散射法)以及ISO133320对分析方法验证内容的具体要求可参见表3。从表中可以看出,USP和EP关于粒度分布方法学验证部分的要求基本相同,明确了精密度中重复性的验证要求,尚未对中间精密度和耐用性的验证要求进行规定。
精密度中的重复性是指实验室内同一操作人员在短时间内使用相同仪器进行验证的过程,中间精密度则是指在同一实验室,由不同实验人员在不同时间使用不同仪器进行测定的过程,进行中间精密度考察时,可考虑参考重复性的验证要求。对于分析方法准确性的评估,可考虑通过测定标准粒子进行仪器校正来证明。
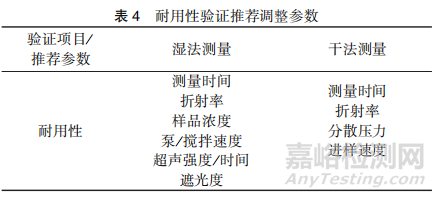
方法的耐用性是指在有意对测试条件进行微小变化后,方法仍能保持不受影响的能力,为日常测试提供正常使用范围的可靠性指标。由于激光衍射法的干法测量和湿法测量的测试参数有所不同,两种方法的耐用性验证内容也有所区别。建议在粒度分析方法开发阶段,对可能影响方法检测的参数进行评估,确定最优参数,并通过调整这些参数的变化来评价方法的耐用性。推荐对表4中列举的参数进行耐用性验证。
目前,《中国药典》2020年版<0982>第三法(光散射法)在测试的相关内容上相对来说比较简单,仅针对干、湿法不同的检测提出了一些建议,由于技术的更新换代,部分内容尚有更新空间。建议参考USP43<429>和EP11.0 2.9.31的要求进行方法验证。药典中的方法验证的接受限度相对较宽,可以根据实际样品调整方法重复性、耐用性要求。
四、如何建立合理的控制标准
应评估粒子或药物颗粒对产品质量的影响,对于被判定为关键质量属性的颗粒,则有必要考虑建立粒度及粒度分布的控制措施,以保证产品质量的稳定性和生产可行性。企业在制定粒度分布控制标准时,通常仅对D50或D90进行控制。单独控制中值粒度或边界粒度不能完整体现颗粒的粒度分布情况,尤其对于粒度分布宽的颗粒而言,应考虑对整体的粒度分布进行控制,即进行D10、D50和D90的控制。此外,企业通常仅设置D50或D90的上限限度,在没有提供充分科学依据的情况下,上限限度可能不能对粒度分布进行有效控制。例如,原料药微粉化技术可显著提高硝苯地平缓释片的体外溶出度,但当D90小于3.471 μm时,制剂出现了突释现象,影响了硝苯地平缓释片的缓释效果[19]。根据粒度分布与生产可行性、产品性能相关性的研究结果,考虑制定D10、D50和D90合理的上限和下限控制限度,有利于为质量控制提供可靠的保障。
五、结语
药物粉体的粒度和粒度分布是影响整个药物制造过程的基础,粒度的特性决定了成品的多种性质。药物粉体的粒度分布可以极大的影响药物的生产可行性(如流动性、压缩性、均匀性、稳定性等)和产品性能(如溶解度、生物利用度等),因此,粒度分布被认为是实现药品目标质量指标(QTPP)的关键物质属性。粒度分布的测量是一个复杂的过程,涉及采样的代表性、分散体系的稳定性、测量参数的合理性等方面。粒度分析方法开发和验证,应基于代表性的样品进行,通过分析颗粒的性质,建立适宜的分散体系并设置合理的测量条件。将QbD方法运用到粒度分布分析方法开发和验证,有助于减少方法的变异性,获得可重复性的测量结果。可考虑对D10、D50和D90分别设置上限和下限来控制粒度分布,更有利于充分监测整体粒度分布的情况,有效保障产品质量。
参考文献
[1] WILLIAMS H D,TREVASKIS N L,CHARMAN S A, alStrategies to address low drug solubility in discovery and development[J].Pharmacol Rev,2013,65( 1) :315--499
[2] BERCUM J. Current events in blend andcontentuniformity[J].Pharm Eng,2014,34(2) : 1-10.
[3] European Medicines Agency.ICH O8: Pharmaceutical Development[ EB/0L].[2023-07-25 ].https://database.ich.orgsites/default/files/08%28R2%29%20Guideline.pdf
[4] POHL M ,SMITH K SCHWEITZER M,et al.lmplications[4]and opportunities of applying ObD principles to analyticalmeasurements[ J].Pharm Technol ,2010 ,34( 2 ) :1
[5] LEANE MPITT K,REYNOLDS G K,et al.Manufacturingclassification system in the real world . factors influencingmanufacturing process choices for filed commercial oralsolid dosage formulations , case studies from industry andconsiderations for continuous processing [ ]. Pharm DevTechnol,2018 ,23 ( 10) :964-977
[6] ROHRS B R.AMIDON G EMEURY R H.et al.ParticleSize Limits to Meet USP Content Uniformity Criteria for Tablets and Capsules[ J].J Phamm Sci-US ,2006,95( 5) :1049-1059.
[7] SHAWN AKAREN M.Investigation of hypromellose particle size effects on drug release from sustained releasehydropphilic matrix tablets [ J]. Drug Dev Ind Pharm ,2007,33(9) :952-958.
[8] FDA.Guide to Inspection of Oral Solid Dosage forms ;Pre/Post Approval Issues for Development and Validation[EB/0L].[ 2023-07-25 ].https ://www.fda.gov/inspec-tions-compliance-enforcement-and-criminal-investiga.tions/inspection - guides/oral - solid - dosage - forms prepost-approval-issues-194#_top.
[9] The United States Pharmacopieial Convention.The UnitedStates Pharmacopeia ( 43th Edition ) : General ChaptersS].2023 :6945,6697,7010.
[10] 国家药品监督管理局药品审评中心.化学药物制剂研究基本技术指导原则[EB/OL].(2005-03-18)[2023-07 - 25 ]. https ://www. cde. org. cn/main/fullsearch/fullsearchpage.
[11] European Medicines Agency. ICH O6A: Specifications :11]Test Procedures and Acceptance Criteria for New DrugSubstances New and Drug Products : Chemical SubstancesEB/0L].[ 2023 - 07 - 25 ]. https://database. ich. orgsites/default/files/06A %20Guideline.pdf.
[12] European Pharmacopoeia Commission.European Pharma-copeia:11.0th Edition[ s].2023 :396-400.
[13] International Organization for Standardization.Particle SizeAnalysis-Laser Diffraction Methods :ISO13320 :2020[ S/OL].[ 2023 - 07 - 25 ]. https ://www. iso. org/standard/691l.tml.
[14] 国家药典委员会.中华人民共和国药典 2020 年版(四部)[S].北京:中国医药科技出版社,2020:145-147.
[15] ANNE VMethod Development for I aser-Diffraction Particle-Size Analysis[J].Pharma Tech ,2010,34(11) :1-7
[16] ALLEN T.Particle Size Measurement ( 4th) [ M].London :Chapman and Hall ,London , 1993
[17] International Organization for Standardization. Particulatematerials Sampling and sample splitting for the determi-nation of particulate properties: ISO 14488 :2007[ S/OL][2023-07-25].https://www.iso.org/standard/39988.ht-m
18] InternationalOrganization for Standardization. Samplepreparation-Dispersingproceduresfor powders inliquids : ISO 14887: 2000 [ S/0L].[ 2023 - 07 - 25 ]https :// www.iso.org/standard/25861.html.[19] 刘洁,方晶,宇阳,等.硝苯地平不同粒度对其缓释片剂质量的影响[J].中国药师,2018,21( 6):1108-1111