摘 要
男性生殖系统用药的生殖毒性评价具有特殊性,需要关注雄性生殖毒性风险,还需关注雄性介导的发育毒性风险。对于男性生殖系统用药,需基于其特殊性,具体问题具体分析,综合受试物的药理/毒理作用机制、同类药物信息、组织分布(精液分布)、适应证特点等因素来开展合适的生殖毒性试验。不同适应证对于生殖毒性风险的接受程度不同,需根据相关试验结果进行获益-风险综合评价,制定相应的风险管控措施,以降低临床试验中受试者和临床应用中患者的生殖毒性风险。本文阐述了国际人用药品注册技术要求协调会和监管机构相关指导原则的相关要求,梳理分析已批准上市男性生殖系统用药的生殖毒性研究情况,并在此基础上提出了男性生殖系统用药生殖毒性评价需要关注的要点。
生殖毒性是指外源性物质产生的对生殖细胞、受孕、妊娠、分娩、哺乳等亲代生殖机能的不良影响,以及对子代胚胎-胎仔发育、出生后发育的不良影响。雄性生殖系统可能受到外界化学物质的干扰而造成损伤,包括睾丸、附睾、精子生成和质量以及激素分泌系统等损伤。药物雄性生殖毒性主要是指药物对雄性生殖系统(睾丸、附睾等)及生殖功能造成损伤,一般包括生殖器官器质性改变、精子数量和/或质量下降、性行为改变、生育力降低、性激素分泌异常等,以及通过精子遗传物质传递,和/或精液-阴道摄入而产生的胚胎-胎仔发育毒性。男性生殖系统用药作为一类特殊治疗领域的药物,直接作用于男性生殖系统,部分品种参与内分泌系统的调节和反馈,而且该类药物一般用药周期较长或可能反复用药,具有较高的生殖毒性风险,尤其是雄性生殖毒性风险,因此需重点关注生殖毒性。本文阐述了国际人用药品注册技术要求协调会( ICH)和药品监管机构相关指导原则的要求,并梳理分析了国内外批准上市男性生殖系统用药的生殖毒性试验研究情况,探讨男性生殖系统用药生殖毒性风险评价的关注要点,以期为该类药物研究与评价提供参考。
1、男性生殖系统用药生殖毒性研究的监管要求
1.1ICH指导原则
ICH M3(R2)《支持药物进行临床试验和上市的非临床安全性研究指导原则》中在第11节中提出了药物生殖毒性试验阶段性研究的一般性要求。针对男性用药人群,ICH M3(R2)中指出“因在重复给药毒性试验中评价了药物对生殖器官的影响,所以在雄性生育力试验完成前,男性受试者可以入选Ⅰ和Ⅱ期临床试验。应在大规模或长期的临床试验(如Ⅲ期)开始前完成雄性生育力试验”,注释2提出了重复给药毒性试验的雄性动物生殖器官评价的要求为:在期限至少2周的重复给药毒性试验(通常为啮齿类动物)中进行睾丸全面标准组织病理学检查。《ICH M3问与答》指出“ICH M3(R2)指导原则并未就男性专用药进行胚胎-胎仔发育试验给予建议。男性专用药的胚胎-胎仔发育试验应遵循具体情况具体分析原则”,指出“在潜在生殖和发育风险明确之前,在男性中采取避孕措施是常规做法”。
ICH S5(R3)《人用药物生殖与发育毒性检测指导原则》为支持药物进行临床试验和上市申请所需的非临床生殖与发育毒性试验(国内称为“生殖毒性试验”)提出了国际协调要求。其中,针对生育力与早期胚胎发育试验(FEED)的策略,指出“当为支持目标人群的暴露需要进行一项FEED试验时,大多数情况下预期包含交配阶段。这种试验通常采用啮齿类动物。如果预期对生育力无不良影响,可以在同一试验中对2种性别进行给药和合笼。如果该试验确定了对生育力有影响,则应确定受影响的性别。相反,如果根据作用机制或重复给药毒性试验的结果预期会有不良影响,则每一给药的单性别可与未给药的对应性别进行合笼。这可以在单个试验中使用单独的给药臂来实现,也可以通过进行2个单独的FEED试验来实现。对生育力和早期胚胎发育不良影响的可逆性评估,对风险评估具有重要影响”。
ICH S5(R3)中针对雄性动物FEED试验设计,指出“对雄性啮齿类动物的FEED试验设计包括合笼前给药2~4周,以使得能检测对精子生成和附睾运输的影响。当重复给药毒性试验资料表明药物对睾丸有毒性时,延长合笼前给药时间至10周是合适的,这使得能评估对完整生精周期和附睾运输的影响。FEED试验还可检测功能性影响(例如性欲、附睾精子成熟、射精),而这些功能性影响不能通过雄性生殖器官的组织学检查来检测”。另外,ICH S5(R3)还指出“当基于药物的作用机制或以往的试验资料对生育力有担忧的理由时,可以在重复给药毒性试验和/或生育力试验中纳入额外的检查(例如,采集精子进行精子计数和形态/活力评估、测量激素水平或监测动情/月经周期),以进一步描述药物对生育力的潜在影响”。
ICH S5(R3)针对生物制品提出特殊考虑。对于生物制品,若在啮齿类动物或兔中有药理学活性,推荐在一种种属中进行FEED试验。在非人灵长类动物(NHP)是唯一的药理学相关种属(如对于许多单抗)情况下,在给药期限至少3个月以上的重复给药毒性试验中对生殖组织进行组织病理学检查可作为生育力评估的一种替代方法。这种方法应包括对雌性和雄性动物的生殖器官进行详细全面的组织病理学检查,且在试验开始时动物应达到性成熟(除非该生物制品拟用于治疗晚期癌症)。ICH S5(R3)在其注释1中提出了重复给药毒性试验中雌雄动物组织病理学检查的要求,其中,对于雄性动物的要求为睾丸和附睾应采用能保存生精上皮组织结构的方法进行取材和处理;对生精周期进行详细的显微镜定性评价是检测对精子发生影响的灵敏方法;尽管一般没有必要,但在试验设计中纳入附加的试验终点(例如免疫组织化学、抗均质化的精子计数、流式细胞术、分期定量分析),可用于进一步表征任何已确证的影响。
1.2美国指导原则
美国食品药品监督管理局(FDA)于2018年10月正式发布了《药物研发过程中睾丸毒性评价指导原则》。该指导原则讨论了关于可能对睾丸产生不良影响(睾丸毒性)的药物的下列主题:①提示药物临床睾丸毒性风险的非临床发现,以及对评估这种风险的程度而可能需要的非临床试验;②当对男性受试者首次给予这些药物时,可采用的临床监测;③主要目的为评估药物相关睾丸毒性的临床试验的设计。
关于非临床评价方面,FDA认为:雄性生殖系统的非临床评价是药物开发过程中非临床安全性评价的一个标准组成部分。动物中对雄性生殖系统的药物相关毒性结果,特别是合适动物种属的雄性生殖器官中药物蓄积证据,确定是否有必要评价药物对男性的睾丸毒性。评估睾丸毒性常规采用以下方法:①2种动物种属给药2~4周的重复给药毒性试验,除非只有一种药理学相关动物种属;②啮齿类动物雄性生育力评估(如适用)。其他信息可来源于胚胎-胎仔生殖发育毒性试验以及出生前、新生仔或幼龄动物暴露后的生育力评估,另外,还应考虑已知的药物类别效应和/或潜在的靶点相关效应。该指导原则还提出的非临床试验设计时的一些考虑点,详见指导原则Ⅲ.B节。
一般来说,引起对生育力损伤担忧的雄性动物的生殖毒性结果包括(但不限于):睾丸萎缩、变性、坏死或细胞数量过少;生精小管变性或坏死增加;生殖细胞耗竭;或者其他可能提示生殖功能受损的病理学变化。此外,在其他相关雄性生殖器官(如前列腺、精囊、附睾)的发现可能提示睾丸毒性。对与生殖功能下降和/或不良组织病理学相关的直接作用的睾丸毒性物质,申请人应考虑睾丸功能的临床评估。
该指导原则中列出了增加雄性生育力担忧的非临床研究的10种结果:①发生于临床相关暴露量或临床暴露量低倍数时的发现;②发生于多个动物种属中的发现。③随着暴露时间延长发生率和严重程度升高的发现;④在末次给药后1至2个生精周期或5个半衰期后,发现未恢复或仅显示部分恢复;⑤在成对器官中双侧均出现的发现;⑥在健康未给药动物中罕见的发现;⑦与不良病理学相关的生殖器官重量改变(升高或降低);⑧雄性生育力降低和交配行为受损;⑨精子质量的不良影响(数量、活力或形态);⑩激素紊乱的表现为抗雄激素表现-雄性性器官重量和成熟降低,包括精囊和前列腺(当其与其分泌物称重),提示攻击性降低的临床症状(例如,嗜睡或交配行为减少,雄性动物雌性化),雄激素体征-雌性动物雄性化(生育力降低,雌性性器官病理,或动情周期),睾丸大小减小,精子发生受损。
该指导原则指出多种因素可混淆明显的雄性生殖毒性,包括体质量下降或损伤神经肌肉/神经功能的药物可能导致与生殖功能受损相符的信号;当在睾丸组织病理学检查中检测到精子发生减少时,记录动物的生殖年龄并确定药物对睾丸发育和精子发生是否具有暂时或永久的影响是很重要的;对大鼠精子质量造成不良影响而不影响交配结果的药物,对人类男性仍然可能是一种风险,因为这些发现可能表明对睾丸功能的不良影响,独立于交配结果。对于其作用机制是基于激素水平变化的睾丸毒性药物,申请人应临床监测激素。
基于对非临床毒理学试验结果和任何其他结果的评估,申请人应基于具体问题具体分析原则,考虑追加非临床研究以表征所观察到的雄性生殖毒性。追加试验可包括以下评估:证明停药后毒性发现可恢复的潜力,若不能从最初的毒理学研究中所获得;生殖激素分析,需认识到激素水平在不同动物间和1天内以及试验过程中可能变异大;靶细胞类型的确定(例如精子细胞、睾丸间质细胞、支持细胞)。在某些情况下,在重复给药毒性试验或生育力试验中纳入生育力和/或精子质量分析可能是合适的。在雄性生育力试验中,可以增加交配前的给药时间,以覆盖整个生精周期和附睾运输(例如,大鼠约为63 d),以确定既往试验中预期或观察到的毒性的程度。在毒性结果被怀疑是种属依赖性时,在第2种种属中的确证性试验是有用的,例如,当毒性结果由种属特异性代谢所致时。
该指导原则提出了对临床试验期间的睾丸监测、评价药物对睾丸影响的临床试验设计的要求,其中包括:根据药物非临床试验结果,预测其在临床相关暴露可能引起人睾丸毒性,应在临床开发早期即制定人睾丸损伤风险最小化和监测计划。对这些要求本文不进行阐述,具体可参见指导原则。
总体上,FDA认为当非临床试验显示对雄性生殖器官、精液分析和/或生育力有药物相关的不良影响时,申请人应考虑其对人类的潜在风险;如果需要对男性睾丸毒性进行评估,应在男性受试者的临床试验的早期,或在药物开发的适当阶段,制定阐述和监测睾丸损伤风险的计划。
FDA于2015年6月发布了《雄性介导的药物发育毒性风险评估指导原则(草案)》。虽然FDA后续未发布该指导原则的正式稿,但其中对于男性生殖系统用药的发育毒性风险评估的科学性和监管理念仍具有借鉴和参考价值。该指导原则(草案)总结了FDA对男性人群使用药物后潜在相关发育毒性风险评估的策略与方法,拓展了对传统雄性生殖与发育毒性的认识与评价。该指导原则(草案)为申请人提供了男性给予活性药物成分(API)后对胚胎/胎仔发育的风险(无论是通过对男性生殖细胞的影响,还是妊娠动物或人类给予显示具有遗传毒性或强发育毒性的药物后经精液转移)评估的建议,该指导原则的详细内容见《FDA〈药物雄性介导发育毒性风险评估指导原则〉介绍》一文。
该指导原则指出:男性在其性伴侣妊娠前/后暴露于API可能会导致其女性伴侣的孕体(胚胎-胎仔)遭受发育毒性风险,男性介导的发育毒性来自妊娠(受精)前药物对精子细胞的损伤或妊娠期间药物通过精液转移至妊娠女性体内(阴道摄入)使孕体获得的直接药物暴露2个方面。当一项临床试验涉及到潜在生殖或发育毒性药物暴露时,风险特征、知情同意、避孕措施是重要的考虑点。研究者在设计包含男性研究对象的临床试验时需要考虑男性受试者给药后对其性伴侣(妊娠或可能妊娠的女性)可能带来潜在的孕体不良影响。鉴于缺乏临床信息,非临床数据将被用于评估风险和为确定关于临床试验期间开展适当预防措施的必要性提供信息。该指导原则提出了雄性介导发育毒性风险评估的考虑要点与建议、与评估药物引起的雄性介导发育毒性相关的非临床研究,也提出临床风险减控的建议(如当预期有风险时,避免妊娠或避免通过精液转移给妊娠的性伴侣)。
该指导原则指出:评价雄性介导的发育毒性风险以制定风险减控策略,依赖于相关的非临床和临床信息的可获得性。评估药物雄性介导的潜在发育毒性时应考虑的重要因素包括①药物或其相关化合物的生殖和发育毒性;②药物的细胞毒性或遗传毒性;③提示毒性风险的药理学作用特征(例如,该药是否直接靶向或间接影响发育信号通路、快速分裂细胞或内分泌功能);④药物的吸收、分布、代谢和排泄特征(例如,在雄性生殖器官中的分布和/或蓄积,或在精液中分布)。基于上述考虑,FDA会综合考虑申请人所提供的全面资料,以支持在临床试验设计时给出是否需要男性避孕的建议,或支持药品上市时的说明书建议。
该指导原则提供了一些与评估药物引起的雄性介导发育毒性相关的非临床研究的内容,体外研究包括遗传毒性标准组合试验、药物对精子(如杀精试验、多种精子遗传学完整性试验)或对胚胎(如胚胎全培养)影响的各种体外试验,体内研究包括成熟雄性动物适当的生殖器官组织病理学检查和/或精子分析的一般毒理学试验、生殖和发育毒性试验标准组合。对于大部分药物而言,评价父代介导的发育毒性仅有的标准体内试验为生育力与早期胚胎发育毒性试验,当仅雄性动物给药时可以进行直接的毒性评价。一旦发现明显的发育毒性影响(如着床前后死亡、早期胚胎畸形),则应开展交配前单性别给药试验,以分析雄性和/或雌性动物单独给药对毒性影响的贡献作用。因标准的生育力与早期胚胎发育毒性试验不能充分发现全部的潜在发育毒性作用,故若雄性单性别给药与未给药雌性交配试验中出现发育毒性信号时,应该考虑开展追加试验,使妊娠动物延长至临产前再行解剖检查。此外,对于动物或人类潜在发育毒性物质,应考虑测定精液中API含量,以努力定量推测可能到达孕体(胚胎-胎仔)中的暴露水平。如果雄性介导发育毒性风险不确定或经检测确定存在,应该对男性研究对象或患者应采取的预防措施类型(如使用避孕套和/或可靠的女性用避孕方法)和时限给予建议,以保证避免女性伴侣妊娠或孕体暴露。
1.3中国指导原则
国内于2006年11月发布的《药物生殖毒性研究技术指导原则》主要参考了起草当时执行的ICH S5(R2)指导原则进行制订,该指导原则对规范和促进我国药物生殖毒性研究起到了重要作用。我国于2017年加入ICH后,ICH指导原则开始在中国实施,因此药物生殖毒性试验研究要求需参考ICH指导原则,如ICH M3(R2)和2020年新发布的ICH S5(R3)等,原2006年版《药物生殖毒性研究技术指导原则》不再适用。
2、男性生殖系统用药生殖毒性研究情况
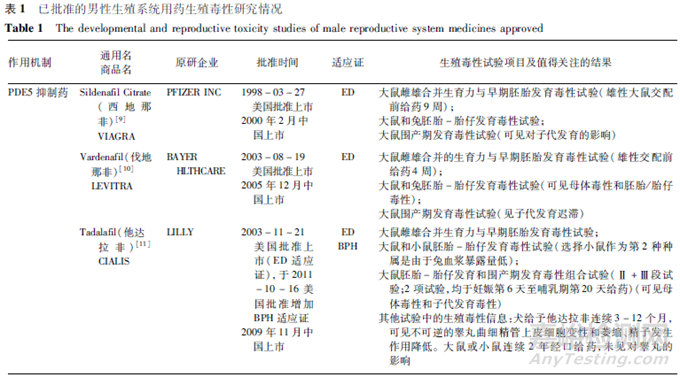
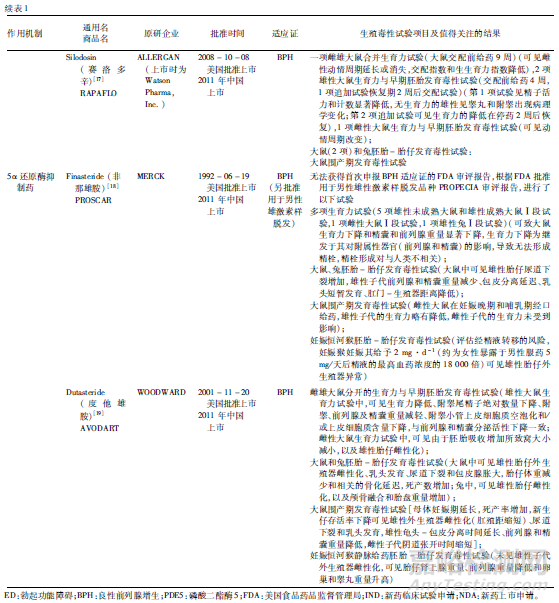
目前获批上市的男性生殖系统用药主要集中于治疗勃起功能障碍(ED)和良性前列腺增生(BPH)。按作用机制分类,主要包括磷酸二酯酶5抑制药(PDE5I)、肾上腺素α1受体拮抗药、5α还原酶抑制药。美国批准上市的男性生殖系统新药中,治疗ED的药物包括西地那非(1998年)、伐地那非(2003年)、阿伐那非(2012年)等,治疗BPH的药物包括特拉唑嗪(1987年)、多沙唑嗪(1990年)、坦索罗辛(1997年)、阿夫唑嗪(2003年)、多沙唑嗪(1990年)、赛洛多辛(2008年)、非那雄胺(1992年)、度他雄胺(2001年)等,获批治疗ED和BPH 2个适应证的药物有他达拉非(2003年)。目前,国内批准开展临床试验的男性生殖系统药物也主要集中于该2类适应证。也有其他适应证品种,例如,用于治疗男性早泄的盐酸达泊西汀片,其作用机制为抑制5-羟色胺再摄取。另外,用于前列腺癌的药物,主要包括去势治疗药物、雄激素拮抗药、化疗药物、个体化治疗药物等,由于其属于抗肿瘤药物,其非临床安全性评价有其特殊性,本文不对该类药物进行讨论。
根据FDA公开的审评意见,对已批准上市的主要男性生殖系统用药的生殖毒性研究情况进行了梳理,汇总见表1。限于篇幅,该表仅列出生殖毒性试验及相关试验中的主要阳性结果,详细结果参见相应品种的FDA药理毒理审评报告和批准的说明书。
分析表1中的品种,上市时间跨度时间较长(1987年至2012年),总体来说上市时间较早,因生殖毒性试验进行时间均早于上市时间,一部分品种的生殖毒性试验在ICH S5首次发布之前(首次的《S5A:药物生殖毒检测指导原则》和《S5B:指导原则附录:雄性生育力毒性》分别发布于1993年6月和1995年11月),也在ICH M3首次发布之前(首次的M3发布于1997年7月),因此,这些品种的生殖毒性研究伴随了国际上对生殖毒性试验的不断认识和完善过程。分析这些品种所进行的生殖毒性试验对男性生殖系统用药研发的参考性,应结合其药物研发具体背景、品种特点,并结合当前国际上对药物生殖毒性和生殖毒性试验的认识进行综合考虑。
分析这些品种所进行的生殖毒性试验,上市前均完成了大鼠生育力与早期胚胎发育毒性试验、2种种属的胚胎-胎仔发育毒性试验和大鼠围产期发育毒性试验,部分品种基于其试验结果或者基于药物所预测的潜在担忧,进行了追加试验。男性生殖系统用药为单性别用药,这些品种采用了大多数药物生殖毒性评价所采用的三段式试验设计,推测其主要原因是,评价药物潜在的生殖和发育风险需要能够评价对生殖过程的所有阶段,若仅进行生育力与早期胚胎发育毒性试验,仅能评价对生育力和早期胚胎发育的影响,无法评价对雄性生殖系统及生殖功能的影响所致的对子代发育的长期影响,因而无法阐述由于雄性/男性用药的全面影响;另外,考虑到男性用药可能存在雄性介导的胚胎-胎仔发育毒性的风险,即精液转移将生殖/发育毒性药物转移至妊娠性伴侣体内,从而导致胚胎-胎仔发育毒性风险,进行胚胎-胎仔发育毒性试验和围产期发育毒性试验,可以考察药物在系统暴露情况下对胚胎-胎仔以及子代生长发育的潜在影响,更好地暴露生殖和发育毒性风险。对于某些类别的药物可能用于多种适应证且包括女性患者,如PDE5抑制药用于肺动脉高压,α1受体拮抗药用于高血压,从药物开发的角度获得全面的生殖毒性试验信息具有必要性。而且,药物上市后,存在妊娠妇女意外暴露的可能,由于药物的生殖毒性的严重性(如致畸性不可逆),从科学性和安全性的角度,识别药物生殖毒性风险并将相关风险写入药品说明书,有助于指导安全用药。但是,对于男性生殖系统用药,尤其是男性专用药,目前,ICH相关指导原则尚未给出具体要求,应遵循具体情况具体分析原则。
从生育力与早期胚胎发育毒性试验看,均进行了雌雄动物生育力试验,大多进行了雌雄合并生育力与早期胚胎发育毒性试验,可能与试验进行时间较早而当时对试验设计的认识有关,但是,在发现问题时追加进行了单性别生育力(尤其是雄性生育力)试验,有些还进行了恢复期观察,以考察生育力影响在停药后是否可恢复。雄性动物交配前给药时间方面,这些品种雄性动物为交配前给药4周或覆盖整个生精周期(大于9周),其中PDE5抑制药除西地那非交配前给药9周给药外,其余均为交配前给药4周,α1受体拮抗药和5α还原酶抑制药雄性给药则均覆盖了整个生精周期。推测原因可能与药物靶点相关风险有关,α1受体拮抗药和5α还原酶抑制药可能对雄性生殖功能或生育力有损伤作用,因此雄性给药覆盖了整个生精周期,而且大部分品种进行了恢复期考察。对于5α还原酶抑制药,根据其靶点和作用机制,预期会对雄性生殖系统和生育力产生不良影响,由于该类药物抑制睾酮向双氢睾酮(DHT)的转化,而DHT是一种雄性外生殖器正常发育必须的性激素,DHT缺乏会导致男性婴儿生殖器的畸形。
对于此类药物,还需重点关注雄性介导的胚胎-胎仔发育毒性。例如度他雄胺,在胚胎-胎仔发育毒性试验中,妊娠大鼠于器官发生期给药,在0.05 mg/次/天(比MRHD低10倍)剂量下可见雄性胎仔外生殖器雌性化(肛殖距减少)、乳头发育、尿道下裂和包皮腺胀大,妊娠兔于器官发生期给药,也可见雄性胎仔雌性化(外生殖器乳头组织学评估),以及颅骨融合和胎盘重量增加。此外,为研究通过精液转移至妊娠性伴侣体内导致胚胎-胎仔发育毒性风险,还开展了妊娠恒河猴静脉给药相当于人类精液中度他雄胺浓度的胚胎-胎仔发育毒性试验,在试验剂量下未见雄性胎仔外生殖器发育的明显异常,但在最高剂量下可见胎仔肾上腺重量、前列腺重量、卵巢和睾丸重量升高。度他雄胺软胶囊说明书提示:妊娠或可能妊娠的妇女不应使用度他雄胺。度他雄胺可经皮肤吸收,可能导致胎儿意外暴露,对男性胎儿有潜在风险。如果妊娠妇女接触到泄漏的胶囊,应立即用肥皂和清水洗涤接触部位。接受度他雄胺治疗的男性在服用最后1剂后至少6个月后才可献血,这一延迟期的目的是防止接受输血的孕妇暴露于度他雄胺。
3、男性生殖系统用药生殖毒性评价的关注要点
基于上述ICH和各监管机构相关指导原则的相关要求,对已批准上市男性生殖系统用药的生殖毒性研究情况进行分析,在此基础上提出了男性生殖系统用药生殖毒性评价需要关注的要点。男性生殖系统用药的作用靶器官为男性生殖系统,需要关注通过雄性生殖系统的影响(如对精子生产的影响)而产生的生殖和发育毒性,影响通过精液转移和阴道摄入转移至妊娠性伴侣而产生的胚胎-胎仔发育毒性,此外,部分品种的适应证可能涉及生育(如ED),更需关注生殖和发育毒性。下面从生殖毒性评价考虑要点,风险识别和控制方面探讨男性生殖系统用药生殖毒性风险评价的考虑。
3.1男性生殖系统用药生殖毒性评价的考虑要点
药物生殖毒性研究最常用的试验方案是三段式试验方案,包括生育力与早期胚胎发育毒性试验(Ⅰ段)、胚胎-胎仔发育毒性试验(Ⅱ段)、围产期毒性试验(Ⅲ段),其中仅Ⅰ段试验涉及到了雄性生殖毒性评价。另外,重复给药毒性试验中对于雄性生殖器官组织病理学检查也是雄性生殖毒性评价的重要内容。然而,正如前文所述,Ⅰ段生殖毒性试验仅能评价对生育力和早期胚胎发育的影响,而无法评价因对雄性生殖系统及生殖功能影响所致的对子代发育的长期影响,从而无法阐述雄性/男性用药所致的全面影响。ICH M3(R2)和ICH S5(R3)等指导原则并未就男性专用药(包括男性生殖系统用药)进行Ⅱ段和Ⅲ段生殖毒性试验给予建议,应遵循具体情况具体分析原则。基于男性生殖系统用药的特殊性,应根据药物的药理/毒理作用机制、同类药物信息、组织分布(精液分布)、适应证特点等综合考虑阶段性开展生殖毒性试验并进行风险评估。
在重复给药毒性试验生殖器官组织病理学检查方面,需要予以高度关注,ICH M3(R2)要求的是标准组织病理学检查,ICH M3(R2)和ICH S5(R3)对组织病理学检查提出的具体要求见上文“1.1”。
在Ⅰ段生殖毒性试验分为2种性别同时给药和单性别单独给药两类设计。对于2种性别同时给药的Ⅰ段试验,雌雄2种双性别动物均给药后交配,可伴随精子分析和组织学检查,评价雄性、雌性生殖性能、生育力和早期胚胎发育毒性。此方法的优点在于减少实验动物使用量、省时省力,对于雄性生殖器官毒性可以给予一定的评价,但是其缺点是如果出现生育力或早期胚胎毒性阳性结果时,无法明确毒性作用是由于雄性给药还是雌性给药造成的,或是两者的联合作用。对于单性别单独给药的Ⅰ段试验,雄性动物给药后与不给药的雌性动物交配,雌性动物给药后与不给药的雄性动物交配,可伴随精子分析和组织学检查,分别评价雄性、雌性的生殖功能、生育力及其胚胎毒性影响。此方法的优点在于可以明确生殖发育毒性中雄性或雌性给药的作用,直接评价药物雄性给药可能产生的生殖和发育毒性。目前国外创新药大多进行单性别单独给药Ⅰ段试验,以获得更多、更直接的雄性或雌性生育力与早期胚胎发育毒性信息;而国内申报资料中的Ⅰ段试验目前仍大多采用2种性别同时给药。
2020年发布的ICH S5(R3)反映了最新的国际协调要求,S5(R3)指出,在预期对生育力无影响的情况下可采用2种性别同时给药的试验,但是若预期有不良影响,则进行单性别单独给药的试验。而对于男性生殖系统用药,因直接作用于男性生殖系统,可能具有较高的生育力风险,因此建议进行单性别单独给药的试验,尤其是雄性单独给药后与未给药雌性进行交配的试验,更直接地评价雄性给药所带来的生殖毒性风险。但是,若综合靶点及同类药物信息而预期无生育力风险,也可考虑进行2种性别同时给药的试验。另外,因标准的Ⅰ段试验不能充分发现全部的潜在发育毒性作用,故若雄性单性别给药与未给药雌性交配试验中出现明显的胚胎发育毒性信号时,应该考虑开展追加试验,如使妊娠动物延长至接近分娩时再行解剖检查。对生育力和早期胚胎发育不良影响的可逆性评估,对风险评估具有重要影响。对于男性生殖系统用药,若常规的Ⅰ段试验发现对雄性生育力和/或生殖系统有不良影响时,应进行追加试验,进行停药后可逆性观察;若在既往的重复给药毒性试验中发现对雄性生殖系统指标有不良影响时,也应研究其可逆性。
ICH S5(R3)指出“如果重复给药毒性试验中发现对睾丸的损伤,在FEED试验中,雄性交配前给药10周覆盖整个生精周期是合适的,这使得能评估对完整生精周期和附睾运输的影响”。“当基于药物的作用机制或以往的试验资料对生育力有担忧的理由时,可以在重复给药毒性试验和/或生育力试验中纳入额外的检查(例如,采集精子进行精子计数和形态/活力评估、测量激素水平或监测动情/月经周期),以进一步描述药物对生育力的潜在影响”。这是对一般药物的要求,对于雄性生殖系统用药,由于其具有较高的生殖毒性风险担忧,建议雄性动物交配前的给药时间足以覆盖整个生精周期,并建议在重复给药毒性试验和/或生育力试验中纳入额外的检测指标。
另外,FDA的《药物研发过程中睾丸毒性评价指导原则》对睾丸毒性的研究提出了较为全面的建议,FDA的《雄性介导的药物发育毒性风险评估指导原则(草案)》提供了一些与评估药物引起的雄性介导发育毒性相关的非临床研究的内容(见上文“1.2”),关于雄性生殖毒性评价指标有文献进行了探讨,可作为男性生殖系统用药非临床研究的参考。
由于男性生殖系统用药具有通过精液转移阴道摄入至妊娠伴侣体内从而具有潜在的胚胎-胎仔发育毒性风险,所以男性生殖系统用药虽然是单性别用药,但开展胚胎-胎仔发育毒性和围产期发育毒性试验可有助于识别和评估风险。
3.2风险识别和控制
风险识别在药物非临床评价中起到至关重要的作用,只有识别了相关风险才能进行后续的风险评估和采取必要的风险管控措施,以保障受试者/患者的安全。对于男性生殖系统用药生殖毒性风险识别主要基于重复给药毒性试验和生殖毒性试验。在新药早期开发阶段,根据非临床研究的阶段性,可能尚未完成生殖毒性试验,重复给药毒性试验中对生殖器官(包括睾丸和附睾)的组织病理学检查是初步识别雄性生殖毒性风险的重要内容。
在临床试验风险管控措施方面,正如《ICH M3问与答》建议,“在潜在生殖和发育风险明确之前,在男性中采取避孕措施是常规做法”。对于男性生殖系统用药的生殖毒性风险识别和评估后,需要对临床试验采取必要的风险管控措施,以确保受试者或其伴侣胚胎/胎仔安全。如果雄性介导发育毒性风险未经识别或确定存在,应该对受试者或患者应采取的预防措施类型(如使用避孕套和/或可靠的女性用避孕方法)和时限给予建议,以避免女性伴侣妊娠或孕体暴露。
男性生殖系统用药生殖毒性风险评价的最终目的是为药物及其拟用的适应证整体获益-风险评估提供依据。对于雄性介导的发育毒性风险,需要通过风险管控措施以最大可能避免造成不良妊娠结果。对于男性生殖毒性的可接受性,需要考虑的因素为该适应证人群的生育需求和雄性生殖毒性的可逆性。
对于男性生殖系统用药生殖毒性风险评价的落脚点为药物及其拟用适应证的获益风险比,考虑不同适应证对于风险的接受程度不同,需在临床开发阶段进行综合评价,降低在后续临床开发阶段的风险。
4、讨 论
目前国内已有一些企业开发的男性生殖系统新药正处于临床开发阶段。对于此类药物,根据具体问题具体分析原则,应根据药物的药理/毒理作用机制、同类药物信息、组织分布(精液分布)、适应证特点等综合考虑阶段性开展生殖毒性试验并进行风险评估。建议重视重复给药毒性试验中对雄性生殖器官的全面组织病理学检查(必要时纳入额外检测指标),并重视其他试验中所获得的雄性生殖系统信息。若在重复给药毒性试验和其他试验中观察到受试物对雄性生殖系统的不良影响,或作用机制或同类品种信息提示可能对雄性生育力有影响时,建议开展单性别给药的生育力试验,且雄性动物给药覆盖整个生精周期;当发现对生育力具有不良影响时,建议追加试验,如设置合适的恢复期以考察毒性的可逆性,进行试验以考察雄性介导的子代发育毒性试验等。根据受试物非临床研究结果,评估其临床相关暴露可能引起生殖毒性风险,在临床试验中应对受试者加强生殖毒性指标监测,并采取严格避孕措施最大可能避免胚胎-胎仔的药物暴露,保护受试者安全。此外,在药物上市时,还需基于非临床和临床试验的所提示的生殖毒性风险信息和相关数据,在说明书中进行提示,包括所需采取的风险控制措施,以降低临床使用中的生殖毒性相关风险。
本文选自:中国临床药理学杂志 第39卷 第20期
作者简介:周植星,副研究员,主要从事新药药理毒理技术审评工作
通信作者:黄芳华,主任药师
作者单位:国家药品监督管理局 药品审评中心