摘 要 / Abstract
目的:依据风险管理理论和相关法规要求,探索行之有效的医疗器械注册自检风险管理模式,以保证其注册产品的安全有效。方法:以注册自检全过程风险管理为研究对象,应用风险要素分析和评定,找出注册自检主要风险环节,进行分析、评价。结果:不同地域、不同岗位人员对注册自检风险管理认识差异较大,其差异性遵循了各地产业发展特点,也导致在技术审评和现场核查中尺度把握的不同。结论:医疗器械注册自检风险管理属于全面质量管理的范畴,应当从监管、企业、检验检测层面多方位将其作为完整的管理体系进行全面策划、实施与风险管理,才能发挥预期作用。
Objective: This study aims to explore an effective risk management model for self-testing in medical device registration based on risk management theory and relevant regulatory requirements. The goal is to ensure the safety and effectiveness of registered products. Methods: The research focuses on the risk management of the whole self-testing process in medical device registration. It employs risk element analysis and evaluation to identify and analyze the key risk points in self-testing. Results: Variations in the understanding of risk management in self-testing for medical device registration exist among individuals from different regions and with different job roles. These differences are influenced by the unique characteristics of industrial development in different places, leading to variations in the scope of technical evaluations and on-site verifications. Conclusion: The risk management of self-testing in medical device registration belongs to the scope of comprehensive quality management. To achieve the desired outcomes, it should be comprehensively planned, implemented, and integrated into a complete management system from regulatory, corporate, and inspection and testing perspectives.
关 键 词 / Key words
医疗器械;注册自检;风险管理;应用研究
medical device; registration self-testing; risk control; applied research
医疗器械注册产品质量保证与其风险管理有着密切的关系。法规要求包含风险管理内容,风险管理理论应用于法规实践与执行的方方面面,融入现代质量管理学理论体系框架内理解,二者为互为表里、相辅相成、互相促进并螺旋推动的关系。2021 年10 月22 日,国家药监局发布《医疗器械注册自检管理规定》[1],作为《医疗器械注册与备案管理办法》《体外诊断试剂注册与备案管理办法》配套文件,允许注册申请人开展注册自检,提交的注册产品检验报告可以是申请人的自检报告,也可以是有资质的医疗器械检验机构出具的检验报告。该规定旨在进一步加快医疗器械的上市进程,继而更好地释放医疗器械产业创新发展活力,同时强化注册申请人的主体责任。这也意味着我国医疗器械监管模式逐步与国际惯例接轨,以促进我国医疗器械产业高质量发展。在我国深化审评审批制度改革、鼓励药品医疗器械创新发展、深化国务院机构改革和职能转变的大背景下,本研究围绕新修订的法规要求,结合目前我国医疗器械法规体系建设现状和医疗器械产业发展需求[2],以风险管理为视角分别从完善配套法规体系、落实监管责任、落实企业主体责任3 个方向深入发掘、分析注册自检制度的实施在加快医疗器械上市进程,释放医疗器械产业创新发展活力,强化注册申请人主体责任要求的实际应用过程中的风险要素,特别是针对过渡期无相应技术审评依据和现场核查标准,以及各地监管人员和企业认识不足的现状所带来的风险提出风险管理模型,并进行定量和定性分析,依据分析评价结果,总结风险存在的规律并深入阐述注册自检风险识别、分析和控制、评价。
1、研究背景、目的及意义
1.1 背景
注册制度改革为企业带来的巨大利好是不言而喻的,同时基于风险出现的普遍规律,应充分认识到政策改革过渡期新增相应的风险也是必然的。这种风险是全方位的,以企业为主体,监管部门政策把握、审评尺度衡量的差异都是风险的重要来源。作为监管部门、技术审评人员、企业三方在共同推动政策改革前行的同时及时做出风险预判并总结风险存在的规律,进行有效地识别、分析和控制、评价有着巨大的现实指导意义。
本课题组进行了大量的文献以及资料的查阅收集,并进行了大量的面向医疗器械生产企业、医疗器械检验检测机构、医疗器械技术审评部门的问卷调查和现场调研。对所有的调查问卷进行了统计和分析,应用了医疗器械风险管理基本理论,制定了以医疗器械注册自检风险定性分析和定量分析为主的研究路径,同时以甘肃省医疗器械生产企业近年来现场检查缺陷项分布情况的量化分析为辅助,采用数据分析法、风险管理工具应用、专家论证和建立企业实践基地的多种结合,对注册自检风险识别、分析、评价、控制以及剩余风险评价进行了全过程研究,从而为相关人员对医疗器械注册自检进行有效的风险管理提供一定的参考。
1.2 目的
依据风险管理基本理论,结合法规要求,以注册自检全过程风险管理为研究对象,应用全过程风险要素分析和评定,找出注册自检主要风险环节,并探索行之有效的风险管理模式,以保证其注册产品的安全有效。
1.3 意义
随着我国医疗器械创新发展进入新的历史时期,对监管科学创新提出更高要求,特别是在医疗器械法规尚不完善的情况下,监管科学与风险管理理论实践的结合将有助于识别内部和外部环境因素可能存在的风险隐患,确认风险隐患发生的概率及危害程度,运用统计学、概率论的方法对风险进行排序,找出存在的主要风险源和分布规律,为下一步的风险处置提供客观依据,从而构建监管与企业双向落实风险防控体系,以预防、承担、规避、转移、转换、控制风险,最大程度地避免、降低和预防可能带来的不利影响,将风险控制在可接受的范围内。以现有法律法规、标准、文件规定与医疗器械风险管理相关标准规定的要求有机融合,将风险管理理论应用于注册(备案)申请人在医疗器械注册自检全过程风险管理,并对识别出的风险点进行研究分析,给出相应的控制要求。将有助于监管人员与申请注册人(备案人)对相关理论的深化理解,指导企业建立有效的风险管理模式并提高实际应用风险管理工具的能力,进一步推动注册自检行为的规范、有序进行。
2、研究方法
2.1 文献查阅
通过收集相关文件资料,经过分析整理,建立医疗器械注册自检风险管理基本理论依据,确定研究过程、主要风险来源和风险管理要素。
2.2 问卷调查
对注册自检过程中所有可能涉及的风险进行识别、梳理,根据风险源、风险发生的可能性和风险发生后的影响程度制定医疗器械注册自检风险要素评定表,并面向医疗器械生产企业、监管部门、检验检测机构、技术审评部门征求意见,对列举的风险来源进行赋值评分。利用风险矩阵法进行数据分析,根据分析结果制定具体的管理措施[3]。
2.3 案例分析
对甘肃省近两年来依据医疗器械GMP 及附录、现场检查指导原则开展的生产质量管理体系核查和注册体系核查主要缺陷分布情况进行统计分析,从而对质量管理风险分布范围界定提供参考。
2.4 理论实践
筛选甘肃省具有代表性的医疗器械生产企业作为项目合作单位,搭建注册自检风险管理实践试点基地。
2.5 专家论证
以现场讨论会(也可为线上会议)的形式向医疗器械监管部门、医疗器械生产企业、行业协会专家广泛征集意见并进行研讨得出结论。该方法是以递进式方式分为多个层级进行,以多方位信息输入的筛选、分析、汇总为基础性方法,通过实践验证和论证结果分析形成最终结果。该方法具有实践性强、可双向反馈的特点。
2.6 研究方法小结
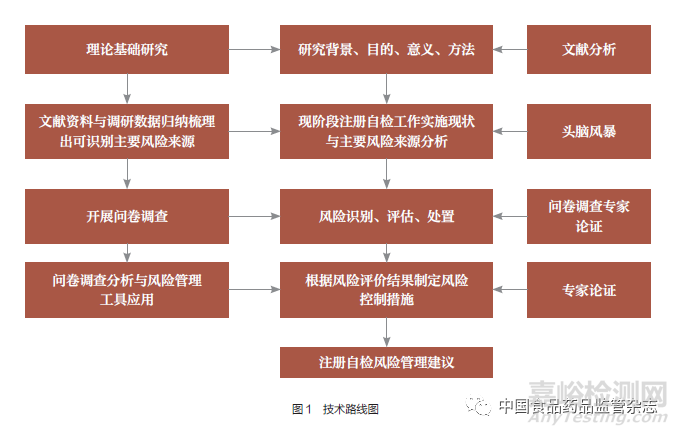
该项目研究内容及技术路线均遵循理论研究与实践数据验证相结合的方法,主要步骤为:一是通过前期大量的文献资料收集和相关国内外法规体系要求的差异分析,同时同步开展调研活动,确定本研究开展的目的、范围、方法。二是通过研究评价目前国际、国内检验检测实验室资质认定评审系统规范、标准体系,结合国务院药品监管部门认定(国家级检验检测机构资质认定)中包含但不限于《检验检测机构资质认定条件》、《医疗器械检验机构资质认定条件》、中国合格评定国家认可委员会认可的规定及相关强制性国家标准、行业推荐性标准体系,以及开展调研意见反馈,通过头脑风暴法梳理集中各类风险要素,确定注册自检主要风险来源。三是依据确定的主要风险来源制定风险评估调查问卷表,设定调查对象及范围,并就问卷形式及内容征求行业内专家意见后随即开展问卷调查。四是对收回调查问卷开展数据统计和分析,通过定量与定性分析,得出风险评价结果后制定风险控制措施,就风险控制有效性组织专家论证会开展剩余风险评价。通过以上研究路线得出研究结论并提出建议(图1)。
3、研究结果
依据《医疗器械监督管理条例》《医疗器械注册与备案管理办法》《体外诊断试剂注册与备案管理办法》《医疗器械注册自检管理规定》《医疗器械生产质量管理规范》法规要求,结合YY/T0287—2017 / ISO 13485:2016《医疗器械 质量管理体系 用于法规的要求》和YY/T 0316—2016/ISO 14971:2007 更正版《医疗器械 风险管理对医疗器械的应用》提供的医疗器械风险管理基本理论,同时参考《医疗器械注册自检管理规定》的具体要求, 并结合CNAS-CL01:2018idt ISO/IEC 17025:2017《 检测和校准实验室能力认可准则》、RB/T 214—2017《检验检测机构资质认定能力评价 检验检测机构通用要求》、RB/T 217—2017《检验检测机构资质认定能力评价医疗器械检验机构要求》,以及2022 年发布的《医疗器械注册质量管理体系核查指南》等规定,设置了调查问卷板块构成,从格式上延续了CNAS-CL01:2018idt ISO/IEC 17025:2017《检测和校准实验室能力认可准则》中列出的控制要素分布。对医疗器械注册自检全过程中风险发生的概率和风险发生后对预期结果的影响严重程度进行定量的表达和评估,以得到科学的风险定量评价模型,用以识别医疗器械注册自检主要风险并研究对应控制措施[4]。基于风险源前后相互关系的交互作用模式,本次调查问卷风险源的征集模型以风险发生的概率和风险发生后对预期结果的影响严重程度为结构,依据本课题组已预估的风险点进行10 分制赋值的方式,被调查者依据主观判定逐项进行评分。基于注册自检活动涉及的风险管理模式、角色、资源、过程及结果应用评价过程的多维度状态,调查问卷主要围绕符合法规要求、基于风险管理的医疗器械质量保障体系为主要视角进行信息采集。因此,除监管部门、专业检验检测机构人员外,将医疗器械生产企业也纳为了调查对象,包括企业研发人员 、质量管理人员、检验检测人员、生产管理人员。根据风险矩阵理论基础,结合故障树分析法(FTA)原理并遵循医疗器械生产质量规范符合性原则,明确目的主线后,将注册自检有效性和符合法规要求设置为顶事件窗[5],以此推断注册自检实施全过程中人、机、料、法、环、测环节可能存在的风险并进行逻辑推理,将故障树中涉及的所有事件以调查问卷形式呈现,并将问卷中各评价指标进行无量纲化计算,即:风险(R)= 风险发生的概率(P)×风险发生后的严重程度(S),得到风险管理研究的综合指数、排序和等级。依据制定的风险管理原则,采取不同的风险控制措施并进行有效性评价[6]。
调查问卷中共归纳出23 个风险环节、66 项注册自检实施过程中可能存在的风险点,利用风险矩阵法对风险可能发生的概率及发生后的严重程度进行定量和定性评价[7],在全国范围选择较有代表性的省市以微信小程序和纸质版形式同时发放调查问卷。共收回问卷368 份,其中有效问卷339 份,问卷有效率为92.1%。监管部门54 份(15.93%), 政府部门下设的医疗器械检验检测机构72 份(21.24%), 医疗器械生产企业213 份(62.83%)。问卷涉及我国广东、山东、上海、重庆、山西、河南、海南、云南、甘肃9 个省份。
3.1 风险评定结果
将企业风险赋值与监管风险赋值进行平均,以保证企业与监管意见的中和。各风险区域分布情况为:风险范围10~17 共分布3 项、风险范围17.1~23 共分布32 项、风险范围23.1~36 共分布31 项,均处于中等风险范围,需采取相应措施将风险降低至可接受水平,并进行剩余风险评价。
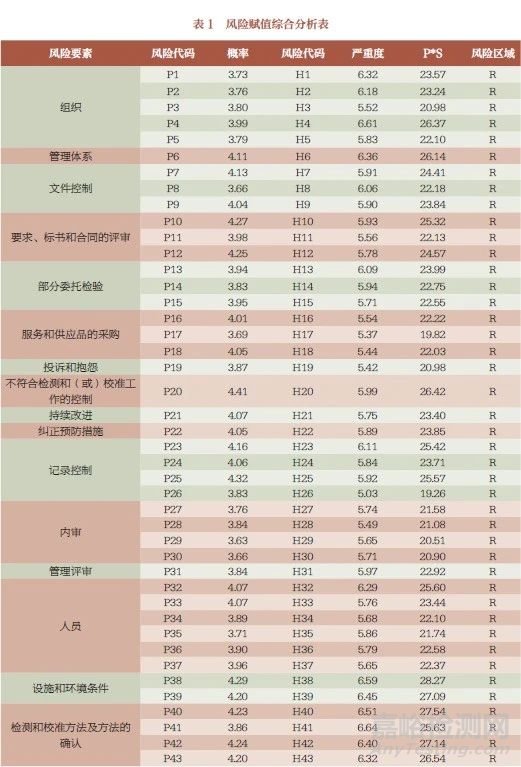
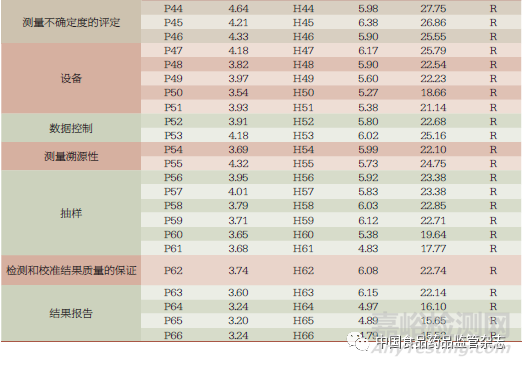
风险指数排名前十的环节依次分布于:设施和环境条件、测量不确定度的评定、检测和校准方法及方法的确认、不符合检测和(或)校准工作的控制、组织、管理体系(表1)。
3.2 风险控制
对以上风险项逐一分别从监管角度和企业发挥主体责任角度进行原因分析并制定风险控制措施后,采用邀请行业内专家评审的方式对剩余风险进行再评价,确定整体风险是否接受[8]。针对采取的风险控制措施,本课题组以行业内具有代表性的医疗器械生产企业作为风险管理措施实践基地。本课题组选取部分行业专家作为风险控制措施有效性再评价方,对提出的控制措施进行再评价,同时由甘肃省医疗器械技术审评部门、检验检测机构、现场检查员同步再评价,整合多方评价要素形成再评价结论,用以评价风险管理的有效性。通过风险控制措施,各剩余风险发生概率和风险发生严重程度显著降低,说明通过风险管理活动,能够有效控制重要风险发生概率。
3.3 监管人员调查数据分析
对调查问卷进行分组排序与统计分析,对收集到的各省、自治区、直辖市监管人员调查数据进行分类统计,对不同省份、不同岗位从事医疗器械检查审评、专职检验、监管执法等人员关于66 项关键风险的发生概率和发生后的严重程度的评估结果进行数据计算分析,通过横向对比得到不同省份监管人员评估差异,通过纵向对比得到不同岗位监管人员评估差异。
经统计,发现各调查省份66项风险评估值最大值与最小值差异比值平均在1 倍以上,反映不同省份不同地区因产业发展特点和监管理念不同,对于同一风险要素的发生概率和发生后的严重程度认知不同,预估值的差异性较大。其中,对风险发生的概率判断,河南与广东评分值相对较高,重庆与云南估值最低;对风险发生后的严重程度估值判断,海南与河南估值较高,山东估值最低。从事医疗器械检查审评、专职检验、监督执法等不同岗位人员对于注册自检风险识别同样存在差异,但其差异性遵循了各地产业发展特点,与其岗位关联程度不高。以上结果反映出各省、自治区、直辖市监管人员对于注册自检风险识别的差异,从而导致在技术审评和现场核查中尺度把握的不同。产业较为发达的省份对特定风险的发生概率与其产业发展是呈正向关联分布的,而对于风险发生后的严重程度认知的不同,更多的来自监管模式与产业发展规模及程度的不同。本课题组在前期与企业开展的调研活动中了解到,涉及监管层面的顾虑与困惑大部分来自企业注册产品在不同省份注册申请时,审评人员与现场检查员在体系核查和资料审查时存在要求不一、结论不一的情况,给企业实施注册自检带来困扰。
3.4 企业人员调查数据分析
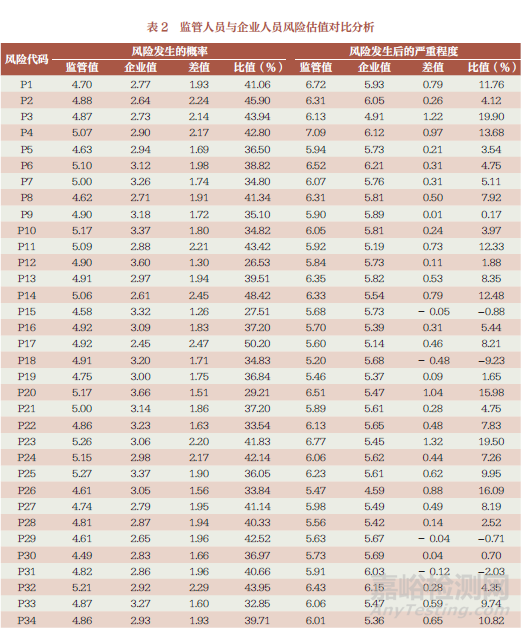
对收集到的来自企业从事研发、生产、质量管理、检验四类岗位管理与工作人员估值信息进行分类统计,发现对于同一风险要素发生概率,企业相关岗位人员估值远低于监管人员,66 项中有24 项风险比值低于监管人员的40% 以上;风险发生后的严重程度估值与监管人员相近(表2)。从岗位分布上看,从事检验岗位人员风险估值普遍高于其他岗位人员。
本课题组对监管人员和企业人员风险平均估值进行了逐一对比和分析,发现风险发生的概率存在较大差异的风险环节主要分布在:组织机构、体系文件管理与法规要求的适用性、采购评审记录、委托检验全过程管理、采购规范性、记录的真实完整可追溯性、内审有效性、管理评审有效性、人员配置与能力验证有效性、检验设备管理、测量溯源性、抽样规范性、注册自检报告规范性。对风险发生后的严重程度存在的差异比值最大为23.42%,主要为关键人员职责、对不符合项的控制和评价、报告中关于型号覆盖说明的要求。
以上分析可以看出,企业人员主观意识风险估值要远低于监管人员估值,由此表明企业方对注册自检风险管理的研究还需进一步加强,而作为监管人员在充分履职尽责的前提下,应当建立综合评判的能力,把握好技术审评工作基本要求和尺度,做到科学界定、重点明确、标准统一。对于列出的66 项风险因素是广泛存在于其体系运行过程和日常生产管理及质量控制中,因此发生概率较高,对于其产生的后果评估是理性且符合企业发展实际的。特别是结合当前“放管服”要求,以鼓励药品医疗器械创新发展为背景,对风险来源的识别既遵循客观事实又能理性分析评判其结果影响是非常必要的。
4、结 论
本研究以医疗器械注册自检政策实施为研究背景,应用全面质量管理理念,建立了风险管理模型围绕注册自检重要的风险环节进行风险管理研究,通过定性和定量分析方法进行了基本的风险等级划分,并进行了可接受风险的排除。通过定量评价的结果得出了各风险点所属风险层级,以及风险分布结论。根据风险评价结果制定了风险控制措施,并对拟实施控制措施后预期剩余风险进行了再评价。在此过程中对注册自检各阶段风险评价结果以及剩余风险进行了简要梳理和说明,通过研究得出如下结论。
医疗器械注册自检风险管理属于全面质量管理的范畴,应当从监管、企业、检验检测层面多方位将其作为完整的管理体系进行全面策划、实施与风险管理,才能更好地控制可能发生的风险。从风险评价结果来看,各方对于注册自检存在明显差异。因此,本着预防为先的理念,各相关方必须建立起全员风险管理意识,将风险管理要求贯穿于整个产品实现的全过程,实施全线管理,多方反馈,以保证产品的风险可控。企业作为主体责任人应当进行充分的风险预判,在不可接受的风险发生前消除隐患,以防风险发生后所带来的巨大损失,从而真正实现注册自检达到预期目的,增强企业发展源动力。
总体来看,监管人员与企业人员对于风险发生的概率与发生后的严重程度认识存在一定的差异。对于风险发生的严重程度评估是理性且符合企业发展实际的,对风险来源的识别是遵循客观事实且理性的。
从法规层面来看,我国医疗器械行业监管法规体系正在不断完善中,配套规章制度逐步完善,其中的风险是可识别且可控的;从企业层面来看,风险管理是一项需长期坚持的工作。监管层对推出的“政策性公共产品”开展理论和实践的风险管理研究,并应用于监管政策具有重大的意义。同时,本次课题研究对于注册自检监管新工具、新方法有效的风险管理具有实践意义。
5、讨论及建议
本研究应用全面质量管理理论基础,将医疗器械注册自检政策实施视为公共“产品”的一种,引入风险管理理念,选择必要的统计学研究方法和风险管理工具,用于注册自检全过程风险识别、评估与处置。医疗器械技术审评机构、核查机构、医疗器械注册申请人可以从以下方面完善监管法规体系和注册自检质量管理体系,以期降低注册自检的风险。
5.1 全面性
医疗器械技术审评工作是在现行法规、标准体系、当前科学认知水平和现有产品技术要求基础上形成的,但随着法规、标准的不断完善,相关科学技术的不断发展,技术审评相关内容及配套文件也需适时进行调整。目前,与注册自检相关联的审评“技术清单”、配套法规、标准体系均有待进一步完善。如何将注册现场核查与注册技术审评要求相关联,以方法和证据提供的科学性、有效性、真实性为重点,开展全面技术审查,保证满足产品技术要求是亟待解决的问题。
5.2 统一性
医疗器械技术审查指导原则是对注册申请人和审查人员的指导性文件,适用于企业准备其医疗器械产品注册申报,同时也适用于审评人员对医疗器械产品上市前申报资料的审查。《医疗器械注册自检管理规定》出台后,由于国家层面尚未出台相应的技术审查指导原则,目前医疗器械审评人员只能依据审评经验评估,因而对技术审评质量的可控性和实效性都形成了一定的制约风险。
5.3 规范性
为确保不同区域、不同机构审评检查尺度的统一,确保医疗器械注册自检政策红利落到实处,一是应尽快研究制定医疗器械注册自检现场检查指导性文件,为检查员开展现场检查提供统一的检查要求,保证不同区域、不同能力、不同机构的检查员保持较为均一的检查尺度,以保证技术审评的稳定性和可重现性。二是尽快研究制定医疗器械注册自检技术指南,为企业顺利开展自检工作提供技术指导。三是医疗器械种类繁杂、学科门类多样、特异性产品多,应借助专业技术机构、医疗器械行业协会、龙头企业,从具体品种入手,建立相关产品的注册自检规范。例如,借助专业技术机构的力量建立体外诊断试剂注册自检规范,借助行业协会的力量建立软性亲水接触镜注册自检规范,借助龙头企业的力量建立重离子治疗系统、人工心脏瓣膜注册自检规范等。
5.4 风险管理应用
风险管理是贯穿于医疗器械全生命周期的活动,其起点是对产品的风险分析,在分析的基础上所采用的设计及制造上的技术措施都是为控制风险[9]。注册检验属于医疗器械从产品设计研发到推广应用过程中的一个重要环节,是为了验证产品临床评价前的安全性和有效性,将产品进行检验得出检验报告的行为,因此也应当遵循YY/T 0316—2016/ISO 14971:2007 更正版《医疗器械 风险管理对医疗器械的应用》。医疗器械生产企业应当将自检纳入风险管理计划,采取有效风险控制措施。2022 年10 月12 日,我国发布医疗器械风险管理标准GB/T 42062—2022 /ISO 14971:2019《医疗器械 风险管理对医疗器械的应用》。该标准等同采用现行有效的ISO14971:2019 标准, 将于2023年11 月11 日实施,实施后申请人势必要调整风险管理策略,更新风险管理文档。监管部门应全面分析注册自检的开展给技术审评及现场核查工作带来的全新考验和风险来源,特别是在过渡期应当将该项工作列入风险排查重点工作目录,制定符合本地区监管实际的注册技术审评和现场核查工作要点或指南。
5.5 真实性
此前,我国医疗器械产品注册需提供有资质的医疗器械检验机构出具的全项目检验报告,转为自检后,企业可以制定适用于自身需求的自检计划进行内部资源合理分配,从而大幅缩减检测周期,加快产品上市进程,自检政策对企业研究成果转化、加快临床应用等具有重要意义,但是其带来的风险也不容忽视,企业期望产品快速完成注册检验,在压缩检测周期的同时,如何保证检测过程合规、真实、完整、可追溯是企业和监管部门需要关注的重点。
引用本文
杨华,李永红,王瑞,张鑫衍,方延学.医疗器械注册自检风险管理的应用研究[J].中国食品药品监管,2023(9):56-67.