无菌隔离器在制药行业的使用愈加广泛,但已有的研究更多地聚焦于设计布局、物料状态等方面,对人员操作因素的影响方面关注较少。以本公司冻干制剂生产车间为例,设计了一系列合理的无菌隔离器内干预操作,并在操作人员实行干预操作的基础上进行气流流型测试和无菌工艺验证。根据验证结果,判定操作人员在日常生产中的操作具有规范性,可为其他车间的无菌隔离器操作人员的培训和考察提供参考。
隔离技术最早起源于第二次世界大战时使用的手套箱技术,用于处理放射性物质以保护操作人员安全 [1]。自 20 世纪 80 年代以来,隔离技术的应用日益广泛。最初,供实验室使用的无菌检查隔离器在欧洲发展起来,该设备通过提供可靠的环境保障来有效防止微生物对待测试物品的污染,可减少假阳性的产生 ;随后,无菌隔离系统开始广泛应用于食品工业、制药工业等领域。目前,国际上常用的隔离系统主要有限制进出屏障系统 (restricted access barrier system,RABS) 和无菌隔离器 (isolator) 等 [2-3]。
相较而言,无菌隔离器的优势明显,因其采用完全密闭的系统将生产空间与周围环境和操作人员完全隔离,可保护产品免遭环境污染 ( 包括来自操作人员在过滤和密封时带来的污染 )。据 2015 年的统计数据,30%的原料药都具有强烈的毒性 [2],无菌隔离器的应用可保护操作人员免受生产过程中有毒、有害物质带来的伤害。此外,无菌隔离器配备的单独高效过滤器、空气处理系统和无菌隔离器集成气化过氧化氢灭菌系统,可自动控制无菌隔离器中的生产环境条件。通过阴性分数法或过度杀灭法证实灭菌隔离器可以使微生物数降至 10–6 [4-6],而在传统洁净室内,微生物数只能控制在 10–3 的水平 [7]。
《中华人民共和国药典》2020 年版 (ChP 2020)四部通则 9206 无菌检查用隔离系统验证指导原则中对无菌检查法等做出了相关要求,并制定了配套的无菌检查用隔离系统验证和应用指导原则,对无菌隔离器的相关验证进行了规范。相关指导原则的出台引起了大量验证技术研究 [8-9]。在对无菌产品的质量研究中,已经确认包括生产区的设计及其设备布局、生产时的环境状况、所有与生产相关的设备及物料的污染状况、人员操作和卫生状况等,每个环节都对最终产品的质量有举足轻重的意义。而已有的研究更多地聚焦于设计布局、物料状态等方面,对人员操作因素的影响方面关注较少。
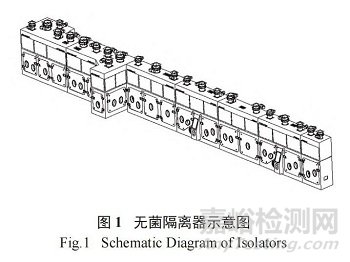
因此,以本公司冻干制剂生产车间为例 ( 图 1为车间使用的无菌隔离器示意图 ),本研究详细论述无菌隔离器内人员操作的规范性对产品质量的重要影响,为其他车间合理使用无菌隔离器、规范人员操作动作等提供参考。
一、无菌隔离器内的操作设计及验证
冻干制剂呈粉状,容易通过呼吸进入人体,从而对人体造成危害。此外,在产品灌装过程中,任何不当操作都有可能导致产品污染。因此本车间对操作人员的动作进行严格规范 :(1) 无菌隔离器中所有动作都不能幅度过大或动作过快 ;(2) 手套不能接触任何与工艺操作无关的表面。上述要求内容均在车间相关标准操作规程中明确,在此基础上,为验证操作人员操作是否符合规范和文件要求,本车间按照相关法规,在确保无菌隔离器内清洁合格、排除前批操作对验证结果影响的前提下,定期进行无菌工艺模拟和气流流型测试。
1、无菌隔离器内的操作设计
要保证操作人员在无菌隔离器内的操作规范性,需先对操作人员在无菌隔离器内的操作内容进行严格设计。设计内容应包含操作人员在无菌隔离器内日常生产时的操作,以及无菌隔离器内设备出现异常时的操作,并确保所有干预操作均是实际生产中可能出现的,不进行不合理干预 [10]。
每批生产均会发生的、正常的、有计划的生产工艺本身固有的干预操作汇总见表 1,包括无菌隔离器内的环境监测、过渡部位对接、在线称重、取样等。纠正性干预操作 ( 即在生产过程中进行纠正或调整的操作 ) 汇总见表 2,包括无菌隔离器使用过程中可能发生的情况,如倒瓶、炸瓶、碎瓶、传感器异常和针头调节等。
2、无菌隔离器内操作规范性的验证
气流流型测试
气流流型测试是指通过将无菌隔离器内的气流可视化,来验证设备运行和人员操作是否会对流型产生影响 [11]。具体测试方法为有资质的验证人员使用烟雾发生器进行测试。在确保无菌隔离器处于关闭状态后,验证人员将设置好的烟雾发生器连接发烟管、调整方向,对准工作区域上方、均流膜下方 30 cm 处,启动烟雾发生器,使烟雾进入气流并随气流流动。
传统的洁净室设计通常在关键区域采用单向流的方式,并且在靠近打开的无菌产品和容器的地方通常要求尘埃粒子符合 A 级洁净度级别,以减少产品和人员污染。本车间根据《医药工业洁净厂房设计规范》和《药品生产质量管理规范 (2010 年修订 )》附录 1 无菌药品的要求 [12-13],A 级洁净区采用单向流。然而,能否控制洁净区内的空气洁净度,气流的影响较大。无菌隔离器内为 A 级环境,能否在日常生产中一直维持其内部的单向流对环境洁净度、产品质量有重要影响。该测试会使用摄像设备全程记录生产过程中的烟雾流动方向。测试过程中,无菌隔离器内设备处于正常生产状态,人员在各个岗位进行模拟干预操作,模拟操作方法见表 1、表 2。通过是否引起紊流或空气停滞等异常状态来验证人员在无菌隔离器内的操作是否规范。
无菌工艺模拟 (aseptic process simulation,APS)
APS 是指使用大豆胰蛋白胨液体培养基来代替药品进行生产 [14]。因为在大多数情况下,微生物在培养基中的生长繁殖要远比在实际产品中容易,所以使用培养基进行生产,其无菌风险远大于正常生产,不仅可用于评价生产工艺对产品无菌保证的有效性和稳定性,还可用于验证人员的无菌操作能力。国家药品监督管理局药品审评中心在对企业的APS 检查中也提出 [10],相关企业在人员的培训与操作方面问题较大。因此,非常有必要对人员在无菌隔离器内的操作进行验证。为保证该验证的普遍适用性,本研究对验证中的人员在无菌隔离器内的操作行动进行规范。在APS 验证中,人员需在无菌隔离器内进行“1.1”项下所述的干预操作,包含生产过程中可能涉及的所有操作,具体操作内容如表 1、表 2 所示。
在整个 APS 验证过程中,按 GMP 要求采用最差的灌装条件,并且对无菌工艺模拟生产的培养基产品进行促生长试验,保证操作人员的任何不规范操作均会反映在培养基产品上。同时,对无菌隔离器内 A 级环境和无菌隔离器所在的 C 级背景环境按正常生产频率进行动态环境监测,监测项目为沉降菌、浮游菌、悬浮粒子和表面微生物。此外,及时记录洁净区之间的压差、温度和湿度,通过环境监测结果和培养基产品的培养结果来验证人员在无菌隔离器内的操作是否规范。
二、无菌隔离器内操作的验证结果
1、 气流流型测试
在无菌隔离器内的设备处于正常生产状态且人员在各个岗位上进行模拟干预操作的条件下,由相关验证人员完成气流流型测试。验证人员应根据实际情况,选择无菌隔离器内的流型测试点,确保人员在无菌隔离器内的模拟干预操作均在烟雾气流下方。气流流型测试动作节选如图 2 所示,人员在无菌隔离器内进行操作时,无菌隔离器内的单向流没有发生改变,未产生紊流现象,且与静态测试相比,气流流型相同,则说明人员在无菌隔离器内的操作规范,不会对无菌隔离器内的环境产生影响,进而影响产品质量。
2、APS 验证
生产前无菌隔离器的准备
无菌隔离器在生产前会进行清洁、消毒、灭菌,以保证设备运行前无微生物污染,不会对验证结果产生影响。
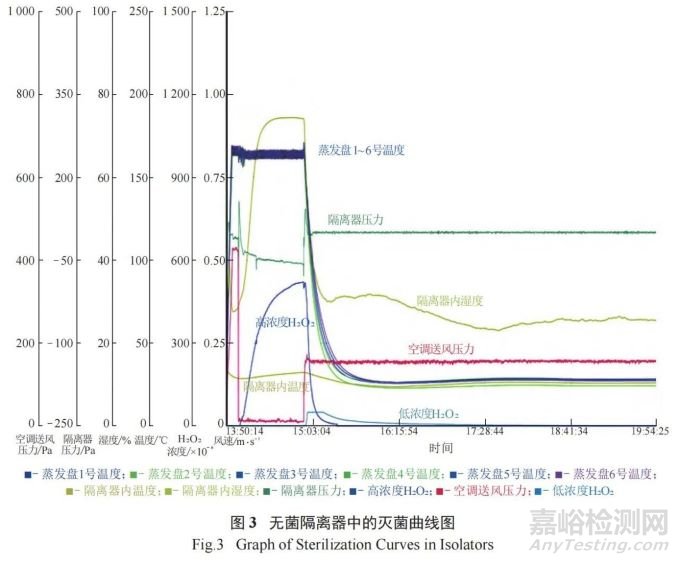
对无菌隔离器内的固定设备和手套等采用异丙醇进行喷雾和擦拭,以对其表面进行清洁和消毒。无菌隔离器内的灌装压塞机需进行泄漏检测,检测合格后,进行在线高温湿热灭菌 (121 ℃,30 min)。随后,将设备运行过程中所需的辅助器具 ( 如不锈钢条和培养皿等物品 ) 经预处理后放置于无菌隔离器中。按无菌隔离器验证的装载示意图定点放置后,对无菌隔离器进行泄漏测试,测试合格后,采用经第三方机构通过对无菌隔离器内的过氧化氢浓度以及生物指示剂的挑战验证得出的灭菌运行参数( 预处理 10 min、充气 15 min、保压 40 min、通风120 min) 进行灭菌,通风至过氧化氢浓度降至 10–6以下,然后对过氧化氢灭菌曲线与岗位标准操作规程模板曲线进行审核,确保灭菌效果达到要求。过氧化氢灭菌曲线见如图 3。
APS
APS 过程在 C 级洁净区背景下的无菌隔离器 A级洁净区环境中进行。操作人员按表 1 和表 2 所示模拟方法进行干预操作,并记录每项干预操作的次数和时间。整个过程通过摄像设备记录,动作节选如图4所示,操作人员通过手套箱操作时,动作干净、移动缓慢且稳健。
APS 过程的环境监测
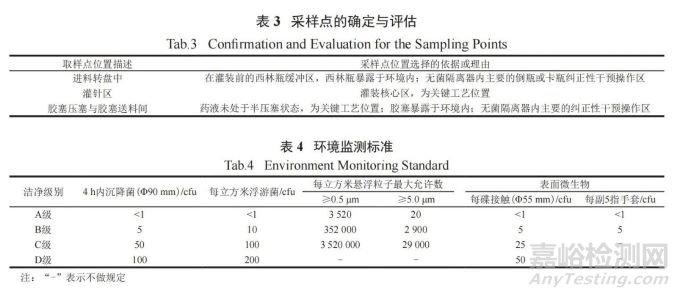
在 APS 全程中进行沉降菌、浮游菌、悬浮粒子和表面微生物的监测。沉降菌、浮游菌和悬浮粒子的采样点确定与评估见表 3,监测标准严格遵守GMP 规定 ( 见表 4)。
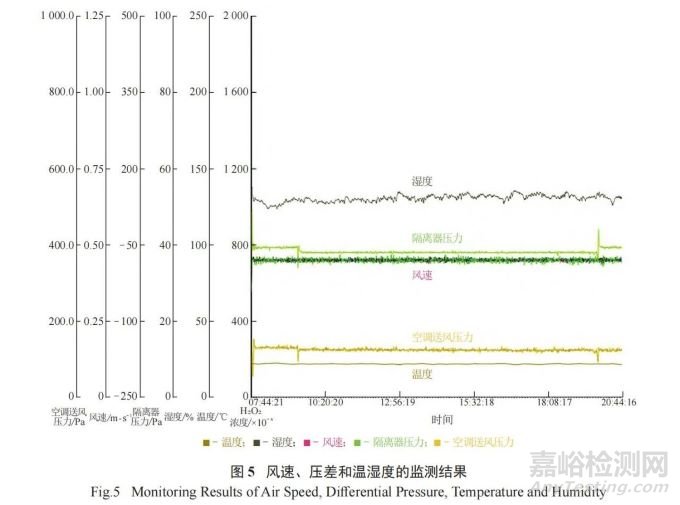
表面微生物的监测除设备表面、无菌隔离器手套表面外,还包括人员监测 ( 如人员穿戴的洁净服表面和人员佩戴的手套表面等 ),进一步对人员在无菌隔离器内的操作规范性进行限制。监测结果表明,整个模拟过程中微生物的污染水平未超过法规规定的限度,证明人员在无菌隔离器内的操作不会对环境造成影响。除此之外,整个APS过程中,风速、压差和温湿度等监测结果均符合相关法规要求,监测记录曲线见图 5。
APS 的培养基产品检查
在 APS 结束后,由车间灯检岗位人员对所有产品进行初步灯检。灯检后,将待培养品充分振摇 ( 确保接触西林瓶内表面 ),然后倒立置于 20 ~25 ℃培养室内培养,培养时间不得少于 7 d ;随后将灌装品翻转,正位放置于 30 ~ 35 ℃培养室内培养,培养时间不得少于 7 d,进行培养观察,并由具有资质的质量管理部人员按 ChP 2020 的要求对培养基产品进行澄清度检查。培养 14 d 后的培养基溶液在澄明度检测仪下仍为澄清溶液,无异物、浑浊,确认无微生物生长。按 GMP 附录无菌药品要求,APS 样品的培养结果应为零污染,因此可根据零污染的培养结果,确认人员在无菌隔离器内操作的规范性。
三、总结与展望
与 RABS 技术相比,无菌隔离器的优势明显。从隔离系统设计本身分析,RABS 的回风系统会产生回风不均匀的问题,导致外围环境空气质量下降 [15],且人员操作的污染风险相对较高。无菌隔离器的背景环境要求在 D 级以上,简化了人员的更衣程序,更方便人员进行生产操作。已有研究对使用过氧化氢进行无菌隔离器灭菌的方法进行了验证,确认其灭菌效果合格 [8—9]。因而,无菌隔离器生产前的灭菌操作可排除其他因素对人员操作验证结果的影响,在此基础上,以实际生产操作为模板设计的模拟干预操作是合理的。
在气流流型测试中,确认静态隔离器内流型为垂直单向,不会对产品产生污染。而在进行生产和人员操作的整个动态过程中,无菌隔离器内风速稳定,且内部的流型恒为单向定流,符合 A 级要求。
整个测试过程中,不产生任何紊流,且无菌隔离器内单向流均匀分布,不会产生气流死角,从而不会对产品产生污染 [16]。在 APS 中,结合灌装产品的培养观察结果和环境监测结果,确认整个 APS 过程中未对产品产生污染。验证结果的合格不仅证明了本车间产品生产工艺的合理性,也表明了无菌隔离器操作人员生产操作的规范性。进行无菌隔离器操作的工作人员在上岗前均已进行过严格培训,使其在无菌隔离器内的操作缓慢、干净,无任何多余小动作 ;即使出现设备故障等异常因素,操作人员进行的纠正性操作也符合要求。
参考文献
[1] 张 秋.隔离器颠覆传统洁净技术——从新版 GMP 对无菌药品的要求看无菌隔离器在制药行业的应用[J].流程工业, 2011, (20): 19.
[2] 李晓雪, 梁 毅.对无菌隔离操作器及其结构与确认的探讨[J].机电信息, 2015, (23): 24-27.
[3] 中华人民共和国国家质量监督检验检疫总局, 中国国家标准化管理委员会.洁净室及相关受控环境 第7部分: 隔离装置(洁净风罩、手套箱、隔离器、微环境): GB/T 25915.7-2010 [S].北京: 中国标准出版社, 2011.
[4] 高海燕, 丁恩峰, 赵丽雅, 等.USP对无菌检验隔离器验证的技术要求[J].医药工程设计, 2010, 31(2): 29-33.
[5] 袁松范.工业指南——用无菌工艺生产的无菌产品-现行GMP(续)[J].医药工程设计, 2005, (5): 44-48.
[6] 袁松范.工业指南 用无菌工艺生产的无菌产品-现行GMP[J].医药工程设计, 2005, (2): 41-46.
[7] 马 伟, 孙石磊.无菌隔离器的优势及其发展趋势[J].机电信息, 2013, (20): 32-36.
[8] 胡桂兰, 岳云燕.无菌隔离系统验证技术的研究[J].轻工科技, 2018, 34(4): 33-34.
[9] 罗辉艳, 严东珍, 刘家媛, 等.无菌隔离器灭菌效果研究[J].中国设备工程, 2017, (6): 78-79.
[10] 胡敬峰, 韩 莹.无菌工艺模拟试验中存在的问题与对策[J].中国药事, 33(12): 1395-1399.
[11] 吴 军.气流保护与无菌工艺操作探讨[J].流程工业, 2018, (12): 44-46.
[12] 中华人民共和国住房和城乡建设部, 国家市场监督管理总局.医药工业洁净厂房设计标准: GB 50457-2019 [S].北京: 中国标准出版社, 2019.
[13] 《〈药品生产质量管理规范〉解读》编委会.《药品生产质量管理规范(2010年修订)》解读[M].北京: 中国医药科技出版社, 2011: 2-5.
[14] 钱静杰.粉针剂无菌工艺验证——培养基模拟灌装试验[J].医药前沿, 2011, 1(20): 217-219.
[15] 钱小进.RABS隔离系统、FFU以及oRABS的取风方式[J].机电信息, 2013, (11): 31-34.
[16] 吴文蕾.无菌检查隔离器的设备与应用要求[J].流程工业, 2017, (6): 28-31.