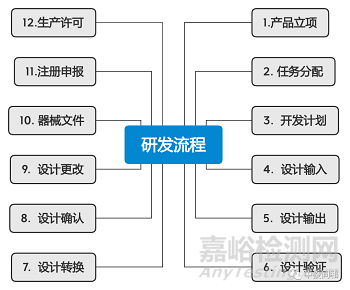
任何产品的研发都是非常严格、漫长而且需要大量资源的投入,医疗器械行业更是如此,因为医疗设备是关系到数百万患者健康的福祉。
医疗器械的开发从来都不是一蹴而就的,通过梳理一下我们汇总整理如下流程,大概需要经过如下12个步骤
中国医疗器械分类
医疗器械是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件。由于不同种类的医疗器械在人体中使用时风险程度具有差异,为了便于监管,我国对医疗器械按照风险程度实行分类。
|
类别 |
定义 |
注册机构 |
|
I类 |
风险程度低、实行常规管理可以保证其安全、有效的医疗器械,如手术刀 |
市级药品监督管理局 |
|
II类 |
具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械,如温度计等 |
省级药品监督管理局 |
|
III类 |
具有较高风险,需要采取特殊措施严格管理以保证其安全、有效的医疗器械,如输血输液器具。 |
国家药品监督管理局 |
美国医疗器械分类
美国医疗器械的监管主要由国家食品与药品监督管理局(FDA)下的器械和辐射健康中心(CDRH)负责。CDRH的主要职能是制定、发布和执行医疗器械标准和质量体系规范,审查和评价医疗器械上市前批准的申请(PMA)和上市前通知(510(k))等。
|
类别 |
定义 |
审查注册方式 |
|
I类 |
风险最低(占47%左右),如医用手套,温度计。 |
一般控制,大部分免于审查 |
|
II类 |
风险比第一类要高(占46%左右),如果失败或,出现问题会对人体或使用者带来不同程度的伤害,但是并不严重,也不会导致人命,如心电图仪,输血输液器具。 |
一般控制和特殊控制,一般需要进行上市前统通知(510k) |
|
III类 |
承担的风险最高(7%左右),多为维持、支持生命或植入体内的器材,对病患具有潜在危险,可能引起伤害或疾病,如心率调节器、子宫内器及婴儿保温箱等。 |
一般控制和特殊控制,一般需要进行上市前通知(PMA) |
一般来说,对于I类产品,企业在向FDA递交相关资料后,FDA只进行公告,不会直接给企业颁发相关证件。但是对于要求严格的对于II和 III类医疗器械,企业必须递交510k(PMN)或PMA,FDA在公告的同时,会给企业以正式的市场准入批准函件(Clearance),此后企业便可以在美国市场销售产品。申请过程中是否到企业进行现场GMP考核,由FDA根据产品风险等级,管理要求和市场反馈等因素综合决定。
上市前通知——510(k) ( PMN: Premarket Notification)
如果所开发的产品是与市场上已有的医疗设备相似的医疗设备,那么510(k)可以作为医疗设备最快推向市场的方法。根据FDA准则的相关规定,在设备上市前至少90天应提交510(k) 。在提交510(k)时,需要说明当前设备与市场上存在的设备存在哪些区别,并能保持相同的安全性和有效性以及预期的用途。尽管需要显示记录在案的实验室测试,但是510(k)提交通常不需要人员数据。但是,如果您提交的510(k)器件已进行了重大更改,则可能需要提供临床试验的结果。由FDA自行决定。关于510(k)途径的另一个重要说明是,它被视为批准程序而非批准程序。这意味着,即使您的510(k)提交内容已被清除,您也不会被广告宣传为“ FDA批准”。
产品上市审核批准——PMA(Premarket Application)
如果您的产品对市场来说是全新的或被归类为III类设备,则将需要申请PMA。如果不能通过采用现阶段已有研究来证明设备的安全性和有效性或被认为具有高风险,则FDA需要广泛的科学证据来确定其是否准备好投放市场。
这通常包括需要提供一定量的实验室和临床试验数据。在申请PMA之前,需要为整个过程中所需要的文件按相关要求做好充分准备,包括临床试验的详细研究计划。FDA保留在此过程中随时批准,拒绝或要求其他数据的权利。因此PMA的成功取得,可以看做是产品拥有了推向市场“护身符”同时是对该类产品本身的最高认可。
中美审批流程差异
1.美国FDA更看重产品本身的危险性,而中国的CFDA则更注重对于管理的难易程度。以输液器具为例,FDA认为器具本身的危险性并不高,因此将其归于II类,而CFDA认为,输液器具会直接侵入人体,若是不严格管理输血输液器具的使用,可能会对人体造成极大危害。造成此类差异的根本在于美国FDA在为器械分类时, 默认要求该器械处于一般控制之下,即默认按照基本管理规范来使用。而我国的CFDA则考虑到管理不当的风险。
2.自2015年7月22日,CFDA发布——国家食品药品监督管理总局关于开展药物临床试验数据自查核查工作的公告(2015年第117号)以来,曾经存在的大量临床试验数据造假现象急剧减少。CFDA希望中国的医疗行业能够重视临床基础数据的重要性,高标准研发,规范生产,因此CFDA的审批越来越严格。
3.根据数据显示
中国:截止2021年1月4日,2020年中国CFDA共批准首次注册三类医疗器械产品1134个,其中国产893个,进口241个,各省级药品监管部门2020年共批准境内第二类医疗器械注册14482个,一类备案医疗器械30739个。
美国:截止2021年1月4日,2020年美国FDA共批准2906个510(k)途径的产品,其中一类器械88个,二类器械2751个,三类器械1个,未分类66个。2020年共有40个通过上市前批准(PMA)途径首次上市。
充分了解中美监管细则的差异及准确评估产品本身的价值,是一个医疗企业所在应该具有的能力,让企业自身产品在最合适的市场快速入场抓住机遇。也是投资人抓住机会发现价值的良好契机。知己知彼,方能百战不殆。