您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-11-26 14:19
元素杂质又称重金属,重金属原义指比重大于5的金属,元素杂质包括可能存在于原料、辅料或制剂中,来源于合成中催化剂残留、药品生产制备过程中引入或辅料中存在的、生产设备引入、或容器密闭系统引入。某些元素杂质不仅对药品的稳定性、保质期产生不利影响,还可能因为潜在的毒性引发药物副反应。
因此欧盟、美国对杂质的控制越来越严格,对此项不断修订,中国在加入ICH后对此项检测应该也会向国际靠拢,因此了解法规对元素杂质的要求、建立有效的检测方法变得尤为重要。
一 USP对元素杂质的修订历程
FDA规定在2018年1月1日之后,针对USP药典品种,提交新的NDA、ANDA应该符合USP<232>、<233>。针对非USP药典品种,申请人提交新的NDA、ANDA时,应该遵循Q3D。美国对元素杂质的规定与ICH规定在不同时期,内容不一致,但从2017年12月之后,USP对元素种类和限量均与ICH保持一致。修订历程详见下表。
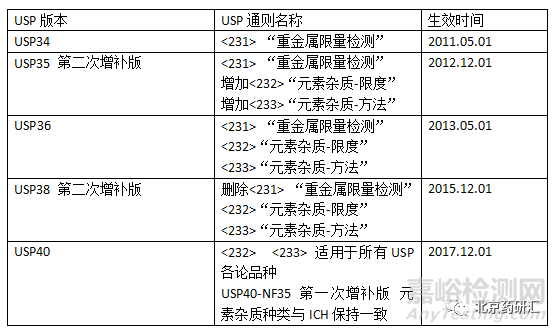
二 哪些产品需要控制?
本指南适用于化药制剂产品以及生物药产品,包括含有纯化后的蛋白质和多肽(包括采用复合或非复合来源生产的蛋白质和多肽)的药品及其衍生物、合成多肽、多核苷酸和低聚糖的药品。
不适用的药品包括不适用于临床研究阶段的药品,也不适用于植物药、放射药、疫苗细胞、代谢产品、DNA产品、血制品、渗析溶液、产品中元素有疗效的药品,还有用于基因治疗、细胞治疗、组织再生的药品以及保健品、兽药、全静脉营养液。
三 元素杂质内容简介
因为目前美国和欧洲的指导原则,美国和欧洲的药典对元素杂质的要求,基本和ICH保持一致。ICH对元素杂质要求有很多专家做过详细的分析,所以本文以ICH对元素杂质要求做简单的分析。
ICH对元素杂质的分类,如下:
Q3D元素杂质指南为口服、注射和吸入制剂中的24个元素杂质建立了允许日暴露量(Permitted Daily Exposure,简称PDE)
第1类
元素砷(As)、镉(Cd)、汞(Hg)和铅(Pb)是对人有毒性的物质,药品生产中不得使用或限制使用,通常来源于矿物赋形剂。因此,所有给药途径的风险评估中都必须评价这4种元素。
第2类
本类别中的元素一般被认为是与药物的给药途径有关的物质,又分A,B两类。
2A类:元素钴(Co)、镍(Ni)、钒(V)。在药品中出现可能性相对较高的元素,因而需要对所有元素杂质的潜在来源及所有摄入途径(如所指)进行风险评估。
2B类:元素银(Ag)、金(Au)、铱(Ir)、锇(Os)、鈀(Pd)、铂 (Pt)、銠(Rh)、钌(Ru)、硒(Se)、铊(Tl),这些元素在自然界中稀少,在药品中出现的可能性较低。除非其在原料药、辅料或药品的其它成分生产中被有意加入,否则可被排除在风险评估以外。
第3类
这类元素包括钡(Ba)、铬(Cr)、铜(Cu)、锂(Li)、钼(Mo)、锑(Sb)、锡(Sn)。口服途径给药,除非在原料药、赋形剂或药品的其他组分生产中有意添加,否则不需要在风险评估中进行考虑。对于生产中非有意添加的这些元素,锂、锑和铜在注射和吸入给药时需进行风险评估;钡、铬 、钼和锡仅需在吸入给药时进行评价。
其他元素:
由于固有毒性低和区域法规差异,没有规定PDE,在该指南中未予以阐述。如果这些元素出现在药品中,按照其他指南或区域法规进行处理。该类元素包括铝(AL)、硼(B)、钙(Ca)、铁(Fe)、钾(K)、镁(Mg)、锰(Mn)、钠(Na)、钨(W)、锌(Zn)。
因为美国药典旧版出现过元素杂质种类、限度和ICH不一致的情况,如表4,很多相关分析文章对此进行了详细的比较。 但是,目前新版USP的规定变更为和ICH一致,该情况应该引起注意。新版USP和ICH对规定元素种类和限度如表5(部分内容,具体规定参见ICH Q3D)。
三 无机元素杂质必须定入产品质量标准吗?
PDE值的30%用来衡量是否将无机元素杂质定入药品的质量标准。如果药品中所有来源的元素杂质水平低于PDE值的30%水平,而申报人已对数据进行了适当的评估,证明对元素杂质的控制已经足够充分,则不需要制定质量标准。
如果风险评估证明元素杂质的水平高于30%PDE,则要建立质量标准来保证元素杂质水平不会超过药品的PDE值。
在提交申报时,如果没有其它论证,一种元素杂质的水平和可变性可以通过提供成分或药品具代表性的、3批生产规模或6批中试规模批次所得的数据来建立。对于有些具有内在可变性的成分(例如,矿物质辅料),在应用控制阈值时,除了30%的PDE值,可能还需要提供额外的数据。
四 药品的元素杂质检测法
采用USP<233>或自己验证过的分析方法来检测成品中无机元素杂质的含量,来评估成品中的各个无机元素是否满足USP/ICH的PDE限度。
每日PDE≥测量值(ug/g)X每天最大剂量(g/day)
这个方法的主要优点是直观,并考虑到了潜在的工艺设备产生的金属污染。
五 元素杂质的计算方法
1 总量计算法
采用USP<233>或自己验证过的分析方法来检测各个原辅料组分中无机元素杂质的含量,按照每个组分的实际用量和药品的最大日用量进行计算,来评估成品中的各个无机元素是否满足USP/ICH的PDE限度。
其中:M=制剂中的每个组分
CM=该组分中重金属浓度测量值(µg/g)
WM=制剂中组分的重量(g)
DD=每日给药最大剂量(g/day)
2 单组分评估法
对药品每日剂量不超过10g的品种,如果在制剂中每个原辅料组分的重金属浓度不超过USP<232>中的表3或者ICH Q3D的表A.2.2中的浓度限度,说明成品中的各个无机元素满足USP/ICH的PDE限度,不需要进一步计算。
方法1和方法2的优缺点如下:
主要优点:
(1)单组分样品处理比制剂样品处理更简单;
(2)可以避免单个组分重金属超标导致成品元素杂质超标;
(3)单一组分的测定结果可以用于多个制剂品种的计算。
主要缺点:
(1)需要对成品中每个组分进行检测,分析工作量巨大;
(2)如果单一组分有多个供应商,需要对每个供应商提供的该组分进行检测;
(3)没有考虑到潜在的工艺设备产生的金属污染。
六 结语
如何评估内包材中的无机元素杂质对液体和半固体制剂的影响?从内包材中可浸出的(leached)元素杂质:对可能从内包材引入的潜在元素杂质应基于药品类型和其它包装材料间的可能的相互作用进行评估。如果通过对结构材料的审核证明内包材不含有任何元素杂质,则不需要进行额外的风险评估。
由于无机元素从内包材浸出至固体剂型的可能性是非常小的,不需要在风险评估中进行深入考虑。但对于液体和半固体剂型,在药品的货架期内,元素杂质从内包材中浸出到药品中的可能性比较大,此时应对内包材中可能浸出(leached)的元素杂质进行研究(在清洁、灭菌、辐射后等),一般要在药品的内包材评估中对该类元素杂质做重点论述。
总之,无论是FDA、USP还是ICH都已经明确的要求从2018年1月1日起对所有上市的、正在申报的、将要申报的药品进行无机元素杂质的风险评估,来保证药品的安全性。

来源:Internet


