您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-08-14 21:51
摘 要 Abstract
《药品说明书和标签管理规定》《已上市药品临床变更技术指导原则(征求意见稿)》《药品上市后变更管理办法(试行)》等法律文件的颁布标志着我国药品说明书上市后变更体系的建设迈上了一个新的台阶。但是,在全生命周期监管的视角下,药品监管部门该如何收集来自被动监测系统如企业、患者以及主动监测系统如哨点医院等多来源的信息,跨部门、跨专业统一并协调药品说明书的上市后变更,成为了新的挑战。本文以说明书撰写、变更所需信息流的管理为切入点,从组织协调、信息来源、变更程序、信息公示等信息流管理视角出发,对欧盟、美国、日本的药品说明书上市后安全性变更管理体系进行综述,以期为我国相关体系的建设提供思路与借鉴。
The promulgation of Measures on Management of Drug Instructions and Labels, Technical Guidelines for Clinical Modification of Marketed Drugs (Draft) and Provisions for Drug Post-Marketing Changes Interim marks a new step in the construction of the post-marketing drug labeling changes system in China. However, from the perspective of full life-cycle supervision, it has become a new challenge for drug regulatory authorities to collect multi-source information collected through passive surveillance system, such as enterprises, patients, and active surveillance system, such as sentinel hospitals and so on, and to unify and coordinate post-marketing drug labeling changes across departments and specialties. In this paper, taking the writing of drug labeling and the management of the changes in the required information flow as the starting point, and from the perspectives of coordination, source of information, the alteration procedures, information publicity, and other information flow management, we review the post-marketing safety labeling changes system in the European Union, the United States and Japan, so as to provide ideas and references for China.
关键词 Key words
药品说明书;多信息来源;上市后变更;启示
drug labeling; multiple sources of information; post-marketing changes; enlightenment
1、背 景
1.1 药品说明书是一份动态变化的协议
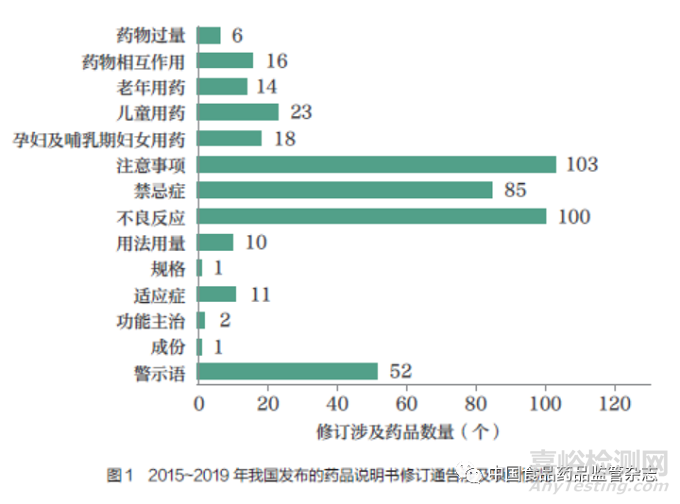
药品说明书是新药研究成果的载体,是用于指导科学、合理用药的法定文件,也是控制药品风险点的重要工具[1]。根据药品品种、规格、适应症等不同,药品可按照处方药和非处方药管理[2],本文主要针对的是处方药药品说明书。一方面,药品说明书是基于以临床试验为主的各种研究证据撰写而成,一定程度上会滞后于临床实践,这是它的先天不足性。另一方面,随着上市后更多的患者被纳入临床实践,更多的安全性和有效性信息被收集,个体化用药经验等得到挖掘,进而有必要对说明书做出一定的变更。近年来,说明书上市后变更已成为我国采取的重要风险控制手段之一,从药品监管部门公示的监管信息来看,根据药品不良反应评估结果要求药品说明书变更是更为常见的形式。说明书上市后变更的内容包括一般性变更,如变更适应症、规格、用法用量,以及安全性变更,如变更不良反应、禁忌症、药物相互作用等。根据2015~2019 年我国发布的药品说明书修订通告涉及项目情况[3]( 图1), 安全性变更是国家药品监管部门发文变更的主要内容。
1.2 国内药品说明书上市后变更现状
1.2.1 法律指南中的变更规定
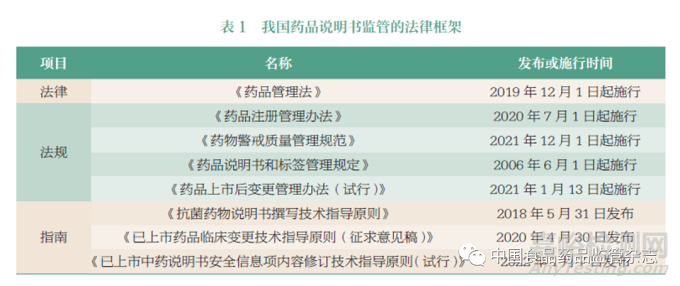
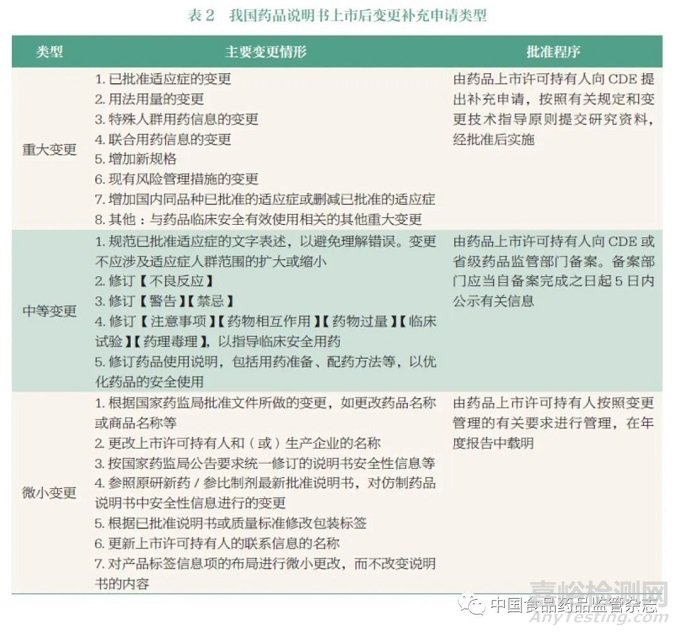
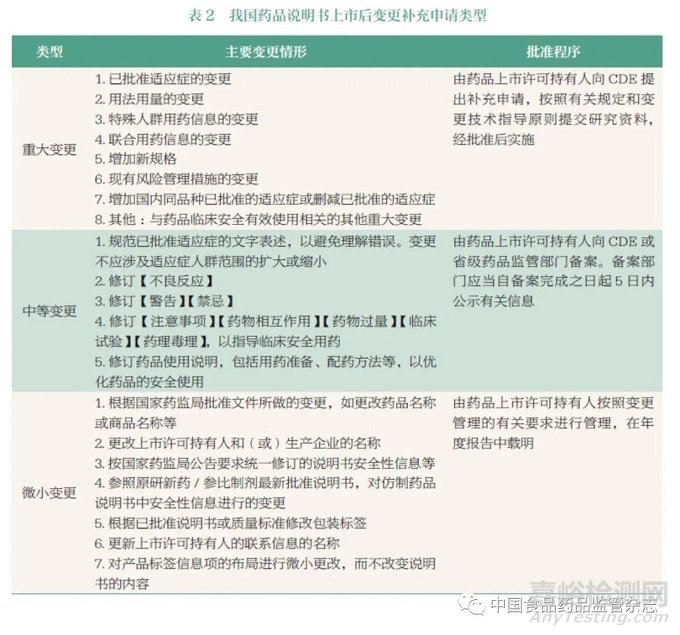
随着《药品管理法》《药品注册管理办法》《药品说明书和标签管理规定》等颁布实施,奠定夯实了我国药品说明书全生命周期监管的法律框架(表1)。在说明书上市后变更方面:《药品注册管理办法》[4] 和《药品说明书和标签管理规定》[5] 都要求申办者应当持续开展药品安全性和有效性研究,及时更新完善药品说明书。同时,药品监管部门也可根据药品不良反应监测和药品上市后评价结果,要求申办者做出说明书的安全性变更。《已上市药品临床变更技术指导原则(征求意见稿)》[6] 中将说明书上市后变更划分成微小变更、中等变更、重大变更3 种类型。《药品上市后变更管理办法(试行)》[7] 则要求申办者应根据上市后药品安全性、有效性和质量可控性的风险大小,主动向国家药品监督管理局药品审评中心(Center for Drug Evaluation, CDE) 递交变更申请,或向地方省级药品监管部门递交备案,或在年度报告中予以载明(具体补充申请类型见表2)。
1.2.2 监管主体与药品说明书上市后安全性变更中的药物警戒
国家药品监督管理局(National Medical Products Administration,NMPA) 是我国药品说明书全生命周期监管的行政主体,在具体的执行中涉及其中多个部门的共同协作。例如,CDE 负责说明书的上市前审批以及上市后重大变更补充申请的批准;国家药品监督管理局药品评价中心负责药品不良反应的常规监测与安全警示;地方药品监管部门负责药品说明书中等变更的备案以及相关行政信息的变更等。
在药品说明书上市后变更的情形中, 虽然我国相关规章指南中规定了药品上市许可持有人(Marketing Authorization Holder,MAH) 负有主动递交药品说明书上市后变更的主体责任,但是在实际操作中,药品监管部门依据药物警戒系统发现的非预期的、严重不良反应,要求MAH 进行安全性变更是更为常见的变更情况。因此,药物警戒系统建设的完善与否,对药品不良反应的评估结果以及说明书上市后的变更情况都起着举足轻重的作用。
目前我国药物警戒支撑系统以被动监测为主, 主动监测为辅。在被动监测方面,已经形成了国家药品不良反应监测中心、省级药品不良反应监测中心、市县级药品不良反应监测中心一体三级的管理体系[8]。医疗机构和MAH 分别通过药品不良反应监测系统和MAH 药品不良反应直接报告系统填写药品不良反应信息。在主动监测方面,原国家食品药品监督管理总局于2016 年成立了国家药品不良反应监测哨点联盟,通过部署中国医院药物警戒系统(Chinese Hospital Pharmacovigilance System,CHPS),辅助医院发现、报告、评价药品不良事件,以获取具有价值的药品安全信息[9]。
1.2.3 信息公示
自2015 年仿制药一致性评价之后,经CDE 审核通过的药品说明书及标签以及上市后变更的说明书电子版本的信息都会被及时公布在CDE 的官方网站上,由于纸质说明书的更新需要一系列流程,CDE 给予其更宽泛的变更时限。此外,国家药品监督管理局药品评价中心与国家药品监督管理局信息中心也会及时公布药品说明书近期的安全警示内容。
总之,我国虽已逐步建立起药品说明书上市后的变更规范,但在具体操作的科学性、系统性、高效性方面仍有需不断完善的地方。与之相比,美欧日在药品说明书上市后变更方面有着多年的实践经验积累,本文借鉴其成功的监管经验,以期为我国药品说明书变更遇到的困难提供解决思路,不断完善我国药品说明书上市后的安全性变更体系。
2、美欧日药品说明书上市后安全性变更体系
2.1 美国食品药品监督管理局(Food and DrugAdministration, FDA)
2.1.1 监管主体与组织协调
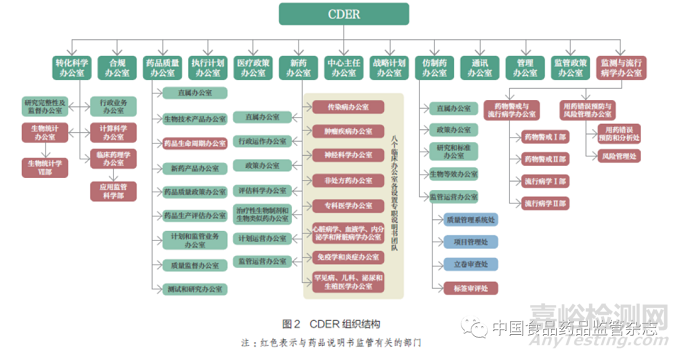
药品审评与研究中心(Center for Drug Evaluationand Research,CDER) 是FDA 负责药品说明书技术审评和监管的主要部门,其下分为超级办公室(Super Office)、二级办公室(Subordinate Office)、部门(Division)和团队(Team),组织结构如图2 所示。其中,创新药和仿制药的说明书分别由新药办公室(Office of New Drug,OND)下的标签专职副主任(Associate Directors for Labeling,ADL)和仿制药办公室(Office of Generic Drug,OGD)下的标签审评处管理,跨适应症的审评会涉及新药办公室下的标签政策协调小组(Labeling Policy Team,LPT);监测与流行病学办公室负责监测、发现、分析、评估安全信号,提高公共卫生水平,建议适当的监管措施,如变更药品说明书[10] ;计算科学办公室会为上市后收集到的风险信息的分析提供创新可靠的解决方法,以提升审查员的工作效率[11] ;药品生命周期办公室负责统一创新药和仿制药的药学质量标准,促进仿制药说明书与其所参照原研药说明书的同步变更, 以实现质量来自于同一个声音(onequality voice)的期望目标,保存并不断完善药品品种档案库[12]。
2.1.2 药品上市后安全性信息来源
在美国,药品上市后安全性信息的来源主要依托于药品不良反应监测系统, 该系统采取被动监测与主动监测相结合的方式。其中,被动监测分为两种上报途径:一是药品生产、经营企业的强制报告系统;二是医疗专家、消费者的MedWatch自愿报告系统。这两个渠道的数据都会汇总到FDA 药品不良事件报告系统(FDA Adverse Event Reporting System ,FAERS)数据库之中。主动监测方面,FDA 于2008 年启动哨点计划[13],旨在通过利用已有的医疗保健数据系统(如电子健康记录系统、报销数据库等)进行上市后主动监测,以补充原有监测体系的不足。FDA 会通过主动风险识别与分析系统(Active RiskIdentification and Analysis,ARIA)进行数据处理、信号挖掘与细化,为监管机构的风险管理措施提供参考决策。
此外, 上市后观察性研究与临床试验、药物性肝损伤监测网络(Drug-induced Liver Injury Network,DILIN)、国家毒理研究中心(National Center for Toxicological Research,NCTR) 和应用监管科学部(Division of Applied Regulatory Science,DARS)所做的毒理学基础研究以及外国监管机构的监管信息等也是重要的安全性信息来源。
2.1.3 责令变更与主动变更
FDA 通过FAERS 数据库、上市后观察性研究与临床试验(自愿或被要求)、哨点系统、DILIN、NCTR 和DARS 的毒理学基础研究以及外国监管机构等多信息来源收集大量不良反应信息和毒理学前沿进展,并利用计算机系统的初次筛选获取可疑的风险信息;接着由监测与流行病学办公室、生物统计办公室、计算科学办公室等进行数据分析与挖掘,进行二次筛选得到修订信号;然后,将修订信号传递给新药办公室与仿制药办公室的说明书专职管理团队,并由其做出说明书是否变更的决定。如果是创新药,则是由新药办公室的标签专职副主任团队和标签政策协调小组进行评议;如果是仿制药,则是由药品生命周期办公室进行管理。但通常情况下,原研药安全性信息来源更为广泛,仿制药与其所参照的原研药保持一致变更即可。最后,CDER 会将药品说明书上市后变更决议传递给MAH,并责令其在规定时限内做出变更,否则,会有极为严厉的处罚措施,同时变更信息也会被传递给说明书数据库的互联网技术管理部门以进行说明书信息更新(具体流程如图3 所示)。此外,在主动变更情形下,申办者主动递交的说明书上市后变更的重大申请,会由新药办公室下属的多学科协调小组进行审评,而对于仿制药的上市后变更是由药品生命周期办公室进行协调。总之,在美国,药品说明书的监管有其专职部门,能够跨部门、跨学科地进行多来源信息的管理(图4),以完善标准化变更流程,并提高工作效率。
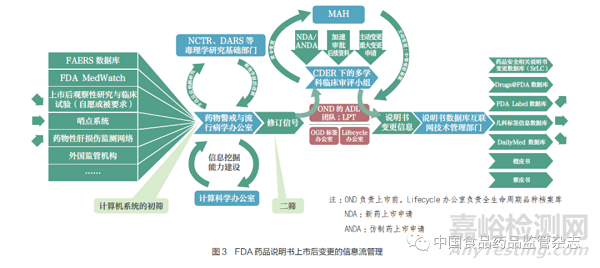
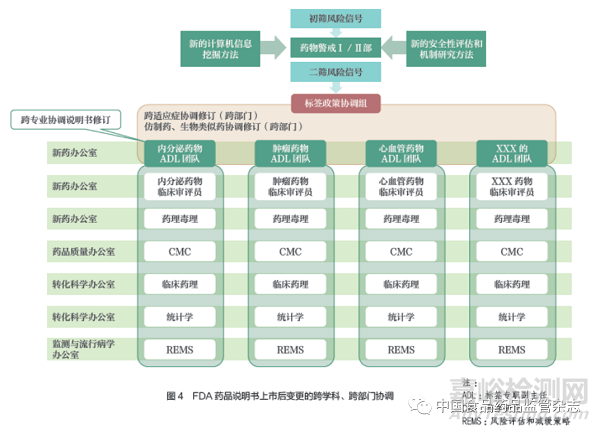
2.1.4 信息公示
在美国, 获批的药品说明书或标签的PDF 文件会在Drugs@FDA 数据库公布。有需要的话,还可以以照片的形式在美国国立医学图书馆维护的DailyMed 数据库中查询到说明书的有关信息。至于药品说明书上市后变更的相应信息会被公布到FDA 官网的药品信息沟通专栏(Drug Safety Communication)以供查询,目前近十年的变更内容都可查询。另外,FDA 对药品说明书信息公示的分类十分细致,细分为药品安全相关说明书变更数据库(SrLC)、FDA Label 数据库、儿科标签信息数据库、橙皮书、紫皮书等,且数据库由专业的互联网技术管理部门进行日常维护。
2.2 欧洲药品管理局(European Management Agency,EMA)
2.2.1 监管主体与组织协调
EMA 是欧盟药品的监管主体,集中与分权是其进行药品管理的基本特征,组织架构如图5所示。EMA 的总体管理机构是管理委员会,负责任命执行理事(Executive Director,ED),执行理事负责EMA 日常事务的管理, 其下设有 6 个事业部。其中,人用药品部门为科学委员会提供秘书服务工作, 而产品特性概述(Summary of Product Characteristic,SmPCs,即药品说明书)的日常管理、仿制药说明书的技术审评与上市后变更同标签办公室、药物警戒办公室、信息管理部门有关。此外,EMA还有7 个委员会(Committees),其中, 负责药品上市后安全管理的是药物警戒风险评估委员会(Pharmacovigilance Risk Assessment Committee ,PRAC),其通常采取主动监测、观察性研究、大型简单临床试验、药物利用研究等进行药物警戒活动[14]。负责人用药品注册管理的是人用药品委员会(Committeefor Medicinal Products for Human Use,CHMP),其会对药品试验数据进行科学、全面的评估,以确保其安全有效,获益大于风险,主要功能集中体现在上市授权与监管科学两个层面[15]。药品说明书上市后的变更依赖PRAC与CHMP 的密切合作,二者相辅相成。
2.2.2 药品上市后安全性信息来源
欧盟有4 种药品审评模式,其中集中审批程序(centralized procedure,CP)是目前最为常见的一种。对于通过集中审批程序成功上市的药品,EMA 的安全性信息来源主要有:欧盟药物警戒数据库(European Union Drug Regulating Authorities Pharmacal Vigilance ,Eudra Vigilance)、上市后观察性研究与临床试验、科学文献出版物、上市后安全性研究、外国监管机构的监管信息等。
2.2.3 安全性变更流程
EMA 药品说明书上市后安全性变更涉及以下几个步骤:首先,PRAC、药物警戒办公室、药物警戒工作组等会收集、汇总、分析、挖掘来自EudraVigilance、上市后观察性研究与临床试验、外国监管机构等多来源的信息,再辅以专业的数据挖掘团队以辅助挖掘安全性信息,期间欧洲毒理学会等基础研究组织也会开展相应的基础研究工作。若PRAC 认为存在有效或潜在的安全信号,会提出相关的药品安全性建议[16] ;接着,建议会递交给CHMP 进一步评议是否需要采取监管措施,如变更SmPCs 等, 如果CHMP 确定要变更药品说明书,则会由执行理事下的标签办公室告知MAH,要求其在规定时限内变更药品说明书;最后,药品说明书变更信息会被传递给EMA 的数据维护部门[17],以更新欧盟公众评估报告数据库(European Public Assessment Report,EPAR)等,并给医疗保健人员提供及时的安全性邮件服务,保障药品说明书信息的及时更新。
2.2.4 信息公示
对于药品的安全性建议方面,有些关键建议或高水平成果会在PRAC 会议之后,CHMP 会议之前,在欧洲药品门户网站上予以及时公布,而完整的科学建议会与CHMP 程序的最终结果一起发布在欧洲药品门户网站上。
对于欧盟上市药品说明书信息,EMA 会在删除商业机密资料之后立即在EPAR 数据库中公布,并提供相关成员国语言的翻译版本。且当SmPCs 做出变更时,其相关文件也会在数据库中及时更新。与此同时,说明书信息在相关成员国主管当局的网站上也可查阅。另外,根据《良好药物警戒规范指南》(Guidelineon Good Pharmacovigilance Practices,GVP) 的安全沟通模块,MAH 应向医疗保健人员及时传播单一的直接医疗专业沟通(direct healthcare professional communication,DHPC),确保其获得第一手的资讯。EMA 还会以药品安全通讯(Newsletters)的形式向患者、消费者和医疗保健人员提供人用药品的关键信息,且每月定期更新。
2.3 日本药品及医疗器械综合机构(Pharmaceuticals and Medical Devices Agency,PMDA)
2.3.1 监管主体与组织协调
PMDA 致力于确保药品从研发到上市后阶段的质量、安全性和有效性,是日本药品与医疗器械产品的技术监管机构,主要包含审评、安全、救济三方面工作的职能部门,与药品说明书上市后变更有关的组织结构如图6 所示。其中,新药Ⅰ~Ⅴ部办公室同时负责新药上市审查和上市后再审查。药物警戒部门负责药品上市后的管理,信息和安全管理办公室负责说明书数据库的信息更新。另外,为了保障风险信息收集、处理的连续性与协调的及时性,部分专业人员会兼任新药Ⅰ~ Ⅴ部办公室和药物警戒Ⅱ部办公室的职责[18]。
2.3.2 药品上市后安全性信息来源
日本的药品上市后安全性信息来源众多:早在2001 年,日本就设立上市后早期阶段警戒(early post-marketing phase vigilance,EPPV) 制度, 要求MAH 负责新药上市后6 个月内所有的药品不良反应并实施安全措施;2013 年,日本实施药品风险管理计划(Risk Management Plan,RMP), 要求MAH 在半年或一年内提交药品安全性定期报告或自愿报告;日本药物警戒数据库(Japanese Adverse Drug Event Report , JADER)[19]会收集来自消费者、医务人员、制药企业的不良事件报告,逐步形成了政府- 药企- 社会公众治理链[20]。此外,还有再审查系统、再评价系统、科学文献出版物等渠道的安全性信息。
2.3.3 安全性变更流程
一般来说,药物警戒Ⅰ部办公室负责药品不良反应监测,收集来自JADER、EPPV、RMP、再审查系统、再评价系统、科学文献出版物、外国监管机构的经验等多来源信息,在积累一定数量信息的基础上,由药物警戒Ⅱ部办公室负责进行每日例行评估与风险彻查,例如会从以集成电子病例为基础的医学信息数据库网络(MID-NET.) 中提取数据,使用药物流行病学方法进行数据挖掘分析。药物警戒Ⅱ部办公室还会统一收集国际人用药品注册技术协调会(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)等国际性信息,每周进行2 次信息筛选,基于流行病学方法,并辅以专业学者的咨询,科学评价收集到的信息,并制定相应的安全对策,如药品说明书变更等[21]。最后,PMDA 将变更的通知告知MAH,要求其变更药品说明书,并由信息和安全管理办公室更新IyakuSearch 数据库等。
2.3.4 信息公示
为确保产品审查的透明度,PMDA 网站会及时发布药品说明书的内容,向公众提供易于理解的医疗产品安全性信息摘要。例如“患者用药指南”会公示简单易懂的处方药说明和警告,“个人严重药品不良反应管理手册( 面向公众)”概述个人药品不良反应的初期症状,并用通俗易懂的语言阐述早期发现和治疗的要点。此外,PMDA 还提供一项名为“PMDAmedio-navi”的电子邮件信息服务,向订阅该服务的医疗保健专业人员分发重要安全性信息。
2.4 小结
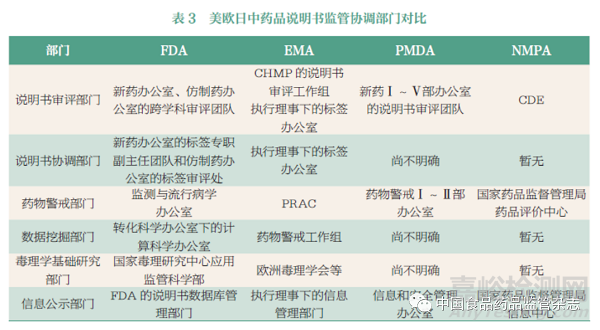
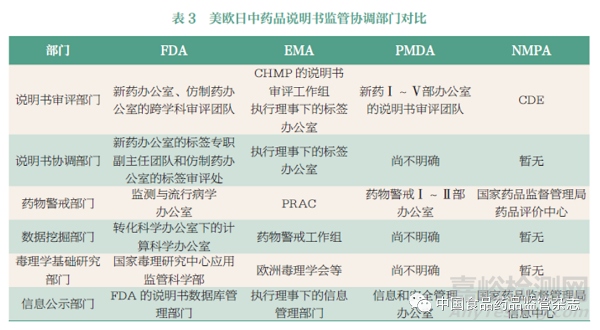
综上所述,FDA、EMA、PMDA、NMPA 在说明书审评部门、说明书协调部门、药物警戒部门、数据挖掘部门、毒理学基础研究部门、信息公示部门方面有着很大的不同,具体对比如表3 所示(由于语言的局限性,日本相关信息有所缺失)。
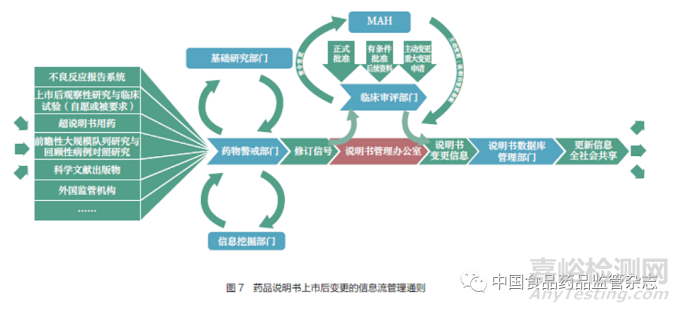
药品上市后的安全性、有效性信息来源众多,安全性信息通常包括上市后前瞻性大规模队列研究、回顾性病例对照研究、不良反应报告、科学文献出版物等,而有效性信息则源于特殊人群使用的剂量调整、提高依从性的处方工艺变更、老药新用的新适应症申请等。普遍而言,美欧日等国家和地区的监管机构都会通过药物警戒系统收集大量的、多来源的信息,在数据挖掘部门的协助下识别安全信号,再由说明书专职管理团队提出安全性建议,以决定是否要进行说明书的上市后变更,并将该要求通知到MAH。最后,变更信息由信息公示部门在数据库中予以更新(图7)。
3、对我国药品说明书上市后安全性变更体系建设的建议
3.1 完善顶层制度设计,强化MAH 主体责任意识,逐步建立以说明书为核心的药品信息监管和指导体系
目前我国对药品说明书虽在法律规章层面予以了规定,也颁布了一系列程序性指导原则,但仅根据药品种类(化学类、生物制品类、中药类)对该类下的整体药品说明书的撰写与变更进行宏观层面的指导,类似于美欧针对具体项目的更为细节上的指导性文件较少,也还缺乏配套的实施机制,普遍存在细节性不够、技术指导性不强、具体变更流程规范不明确、惩处措施不够严肃有力等问题。故建议我国监管机构应随着临床研究、基础研究的进展,数据挖掘等科学技术的进步,不断借鉴国外监管机构成功的实践经验,引入新工具、新方法、新标准,动态更新与完善药品说明书变更的技术性指导原则,明确NMPA 与MAH 双方沟通的步骤、时限和法律责任,必要时可采取财政罚款、资格处罚等措施,以不断强化MAH 在药品说明书上市后变更方面的主体责任意识,化被动为主动,逐步扭转以药品监管部门发文要求变更为主的局面。
3.2 完善药物警戒系统,营造社会共治的良好氛围
我国药品不良反应监测体系趋于被动,主要依赖于国家与地方药品评价中心的不良反应监测系统,且随着上市后药物警戒信息的增加,对数据分析与挖掘工作也提出了更高的要求。因此在不良反应数据积累方面,建议药品监管部门尽可能多地收集药品误用、滥用、过量使用、药物相互作用、缺乏疗效等其他与药品有关的不良事件信息,重点监管疑似严重不良反应的报告,从关注个例报告评价向关注信号转变[22],对严重不良反应或事件所包括的内容做出明确规定,鼓励公众、医护人员及时报告所发现的所有不良事件的情况。通过加强药品上市后安全监管,不断提升药物警戒部门的不良反应监测能力,以倒逼药企更主动地承担说明书上市后变更责任。
另外,建议在收集好的多来源信息的基础上,通过数据挖掘团队进行信号识别、信号验证、信号确认、信号分析及排序、信号的风险获益评估和不合理风险的控制等初筛活动,自动化地挖掘出可疑风险;建议由药物警戒部门建立内部的风险信息编号管理机制,将可疑风险中未解决的难点、痛点进行立卷编号(ticketnumber),随后进行课题立项招标,将课题外包给临床药理、毒理学基础研究实验室、针对性临床观测等专业的第三方组织展开基础研究,得以二次筛选,得出安全信号,并将其交予CDE 做出最终的监管决策,如变更说明书等,达成立卷编号的信息闭环。同时,引入第三方评估手段,在每个财政年,由第三方机构根据相关项目的科技论文发表数量等形式进行药物警戒风险信息编号管理机制的绩效评估,并将评估结果面向社会公开,以供公众了解,逐渐探索药品安全共享共治的和谐氛围。
3.3 在药品审评机构成立说明书协调管理部门,跨学科、跨部门协调,科学规范地撰写和变更药品说明书
对于药品说明书撰写内容与变更内容的审评,本质上是对于庞杂来源的信息流的梳理分析过程,涉及临床医学、药理毒理、药学制造与控制、临床药理、统计学等多个专业的跨学科协同作用;且随着仿制药上市后,对于仿制药说明书与其所参照的原研药说明书保持同步变更的要求也亟待落实。另外,技术性审评的工作离不开事务性工作的辅助,与说明书变更紧密关联的风险信号分析的外包也需要部门协调。从监管主体部门来看,FDA 和EMA 都有着说明书管理的专职团队,能够在相关指南发布、说明书撰写审评、说明书上市后变更、变更信息公示等多个环节起到跨部门协调作用,而我国尚未有专职的负责药品说明书全生命周期监管的部门。故建议可在CDE成立说明书协调管理部门,跨学科、跨部门协调,科学规范地撰写和变更药品说明书,提高审评质量和效率。
3.4 提供药品说明书信息公示平台,构建品种全生命周期档案,打通上市前审评审批和上市后监管的信息孤岛
目前我国药品说明书电子化系统尚待完善,信息公开管理机制有待开发, 尚未建立一个官方的、统一的药品说明书数据库。另外,由CDE 掌握上市前审批的技术数据和由国家药品监督管理局信息中心掌握上市后监管数据之间尚未建立互联互通的信息沟通机制。例如,随着某种药品的仿制药投入市场,当该药品说明书发生变更时,由此引发的是一个类别所有药品的说明书变更,但这样的整体变更情况对于尚未建立品种全生命周期档案的我国来说是非常困难的。因此,建议我国药品监管部门充分利用互联网平台,提供药品说明书信息公示的权威数据库,构建品种全生命周期档案,打通药品上市前审评审批和上市后监管的信息孤岛。

来源:中国食品药品监管杂志


