您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-05-09 07:33
盐酸普萘洛尔原料药和片剂质量标准收载于《中国药典》2020年版(ChP 2020),英国药典2020/欧洲药典10.0(BP 2020/EP 10.0)、美国药典43(USP 43)和日本药典(JP17)也均有收载。其中ChP 2020、BP 2020/EP 10.0、USP 43 和JP17 均采用液相色谱法检查盐酸普萘洛尔原料药有关物质,色谱条件相同。ChP 2020 标准中有关物质采用主成分自身对照法,未对已知杂质进行单独控制,JP 17标准与ChP 2020 限度控制基本相同,而BP 2020/EP 10.0 采用主成分自身对照法,USP 43 采用加校正因子的主成分外标法,对已知杂质A、B、C 进行单独控制。各国药典只有BP 2020/EP 10.0 收载的盐酸普萘洛尔片标准中设定了有关物质检查项,测定方法与原料药相同。
本文在上述标准基础上,对有关物质检查方法进行了改进,改变了色谱条件,采用新型碱性色谱柱系统替代混合离子对方法,优化了有关物质检测波长,筛选了合适的色谱柱并进行方法学验证。同时,对在部分样品中发现的较大的未知杂质进行了分析确证,并综合结构和合成工艺分析该物质的可能来源。
1 盐酸普萘洛尔的有关物质分析
经过对国内企业盐酸普萘洛尔原料药化学反应过程及工艺流程的调研发现,国内企业起始物料、合成路线基本一致,起始物料均为 α- 萘酚(甲萘酚),即现行标准中检查的游离萘酚,合成主要步骤为醚化、胺化和成盐,因而杂质差异不大,所用辅料均为常见的片剂辅料,因此在合成过程中可能存在副产物的产生。此外,盐酸普萘洛尔片生产企业的处方略有不同,但加工工艺基本一致,采用常用的制粒压片法。
BP 2020/EP 10.0和USP 43收录的已知有关物质主要包括杂质A、杂质B 和杂质C,均来源于合成副产物,ChP 2020 检查的游离萘酚,为合成本品的起始物,见表1。
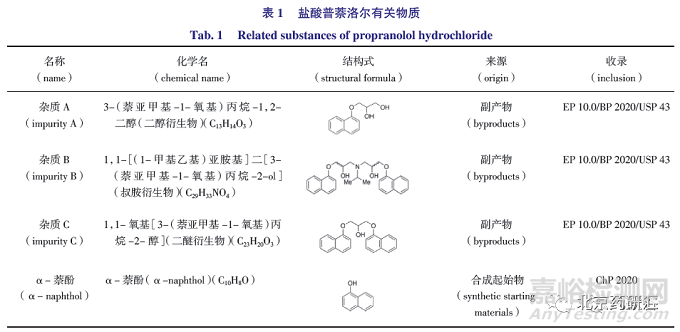
1.1 现行标准方法检验
ChP 2020、BP 2020/EP 10.0、USP 43 和JP17 采用液相色谱法检查本品有关物质,色谱条件相同。分别称取盐酸普萘洛尔对照品(中检院)、杂质A、B、C 和α- 萘酚对照品,用乙腈- 水(50∶50)溶解并稀释制成约含主成分1 mg·mL -1 ,已知杂质均约为1 μg·mL -1 的混合对照品溶液,作为系统适用性溶液(SST)。参照ChP2020 有关物质检测方法,选择Agela Technologies C18(250 mm×4.6 mm,5 μm)色谱柱,以乙腈- 水- 硫酸(55∶45∶0.1)的混合液(取十二烷基硫酸钠 1.6 g 和磷酸二氢四丁基铵 0.31 g溶于1 000 mL 混合液中,用2 mol·L -1 氢氧化钠溶液调节pH 3.3)为流动相,检测波长为292 nm,进样20 μL。结果如图1 所示,混合对照品溶液中各组分峰形和柱效均不理想,α- 萘酚峰与普萘洛尔峰几乎重叠。
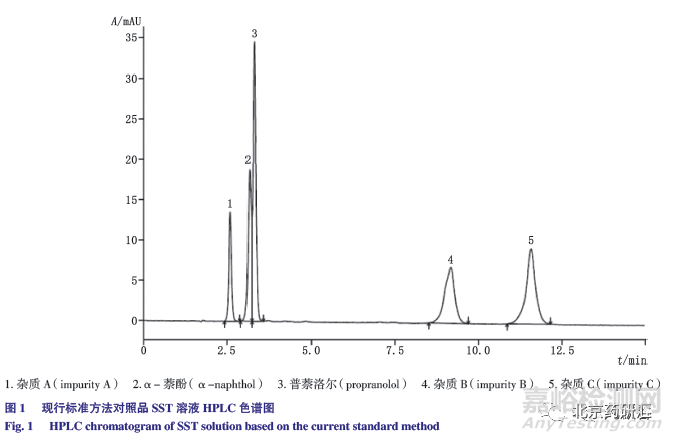
由于α- 萘酚峰是盐酸普萘洛尔在合成中使用的原料,尝试调整有关物质色谱条件,将主成分和各已知杂质在同一色谱系统中实现分离。通过调整十二烷基硫酸钠与磷酸二氢四丁基铵的浓度,以及换用庚烷硫酸钠,均未能达到理想的分离效果,故对有关物质测定色谱条件进行了重新研究,建立了不使用离子对试剂的碱性流动相色谱系统。
1.2 新建方法的色谱条件确定
1.2.1 流动相pH
经软件ChemDraw 预测分析,盐酸普萘洛尔的pKa 为9.5(图2),为兼顾主峰的峰形以及色谱柱的耐受性,采用醋酸铵为缓冲盐,配制10mmol·L -1 醋酸铵溶液,调节流动相中醋酸铵溶液的pH 为10.0、10.5、11.0 和11.3。为进一步提高杂质分离度,改为梯度洗脱。
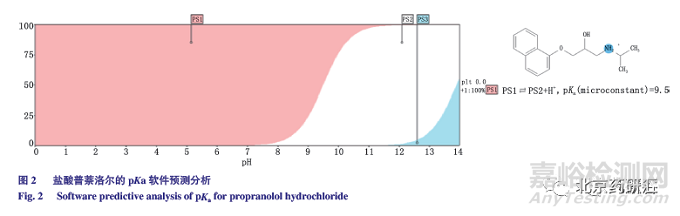
由表2 可知,流动相中水相的pH 对色谱峰的拖尾因子有影响,pH 越低,拖尾因子越大。在该色谱条件下,主峰前发现一未知杂质,与主峰相邻,pH 10和10.5 时的主峰与相邻杂质不能达到基线分离,pH11.3 时的分离度最优,然而pH 过高则对色谱柱的耐碱性要求更高,综合评价pH 11.0 最优。
1.2.2 流动相比例
为进一步提高杂质分离度,色谱条件其他参数不变,仅改变梯度洗脱程序初始流动相的水相和有机相比例,分别设为醋酸铵溶液(10 mmol·L -1 醋酸铵溶液,用浓氨溶液调节pH 至11.0)- 乙腈(90∶10)-乙腈(67∶33)、(70∶30)和(73∶27)。由表3 可知,当比例为73∶27 时,主峰与相邻峰分离度和拖尾因子最好,然而流动相中水相比例增加,主峰保留时间延长,处于梯度变化时间内,有可能影响未知杂质的检出,综合考虑流动相比例确定为70∶30。
1.2.3 色谱柱
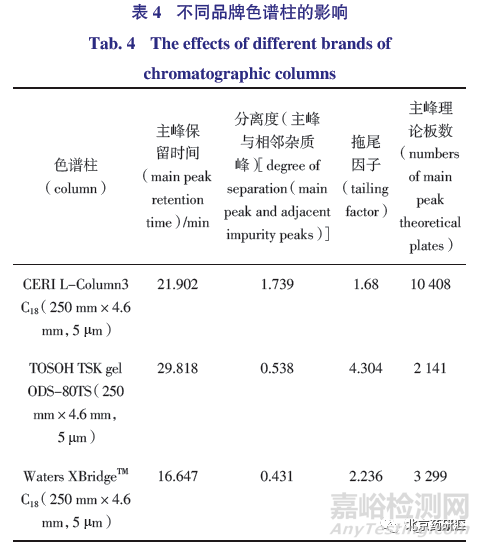
优化后色谱条件使4 种已知杂质和主成分均能有效分离,不同品牌色谱柱对色谱峰的分离度影响比较大,筛选具有高效分离而且耐碱性的色谱柱可能会提高分离度。分别选择CERIL-Column3、TOSOH TSK gel ODS-80TS 和WatersXBridgeTM 的C18 色谱柱,由表4 可知,选择的3 种不同品牌相同规格的色谱柱除与主峰相邻的未知杂质之外,其他杂质均能很好分离。TSK gel ODS-80TS 和XBridgeTM 色谱柱,均不能将主峰与该相邻杂质分离,而TSK gel 色谱柱也延长了主峰的保留时间。经过筛选,CERI 的L-column3 C18 色谱柱能很好地分离相邻杂质和主峰。因此本方法只能选择分离度高的耐碱性色谱柱,推荐CERI L-Column3 色谱柱或效能相当的色谱柱。
1.2.4 检测波长
各国药典标准检测波长均选择292 nm,在上述流动相条件下采用PDA 检测器对SST 中的盐酸普萘洛尔、杂质A、杂质B(显示2 个相连的色谱峰,为同分异构体)、杂质C、α- 萘酚及与主成分的色谱峰相邻未知杂质在190~400 nm 范围内进行光谱扫描。由表5 可知,上述化合物的最大吸收峰集中在213~230 nm 的范围内,292 nm 波长处的吸收均较弱,目标化合物在213~230 nm 的范围内的吸收度远大于292 nm 处,综合考虑梯度洗脱对基线的影响等因素,最终选择230 nm 作为本方法的检测波长。

1.3 色谱条件的确定
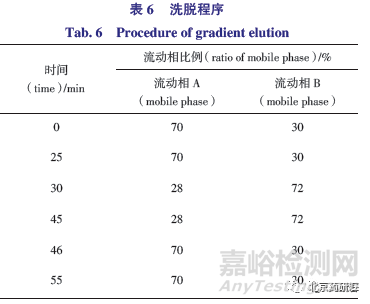
根据上述对色谱条件的探索,确定盐酸普萘洛尔片的有关物质的检测方法如下:色谱柱:CERI L-column3 C18(250 mm×4.6 mm,5 μm);流动相A:醋酸铵溶液(10 mmol·L-1 醋酸铵溶液,用浓氨溶液调节pH 至11.0)- 乙腈(90∶10);流动相B:乙腈;检测波长:230 nm;柱温:30 ℃;流速:1.2 mL·min -1 ;进样体积:20 μL;梯度洗脱程序见表6。
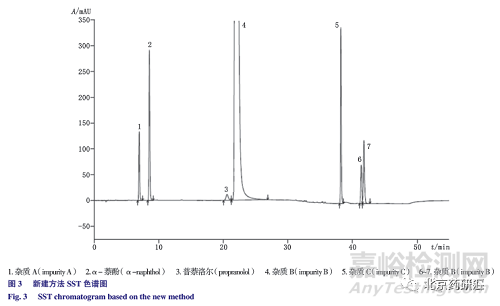
取“ 2.3”项下方法制备ST 溶液,照色谱条件试验,在230 nm 波长进行检测,各已知杂质和主峰相邻未知杂质均可以达到有效分离,见图3。
1.4 与主成分色谱峰相邻未知杂质的结构研究与毒性预测
1.4.1 结构研究
由于所建立的HPLC 检测方法采用了更灵敏的检测波长,且具有更好的分离效率,使得在盐酸普萘洛尔主峰前1 个新未知杂质被检出,该杂质在各国药典中均未提及,但在中检院提供的盐酸普萘洛尔对照品以及部分样品中有检出,且为最大单个杂质,如图4 所示,该未知杂质的最大吸收波长为225 nm,在292 nm 处几乎没有吸收,因此使用现行标准中292 nm 检测波长未能检出该杂质。
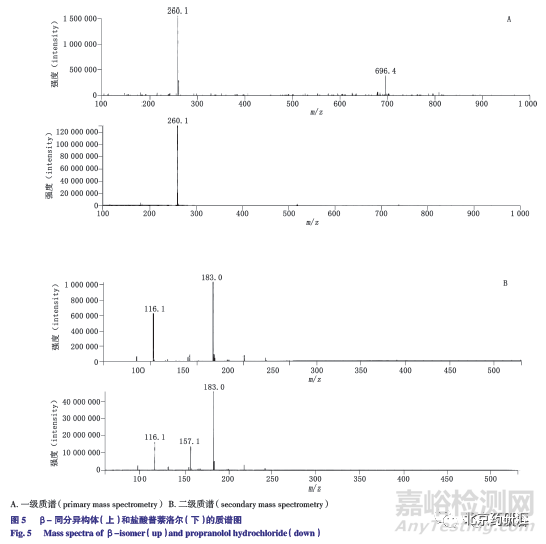
采用上述液相色谱条件,通过质谱检测器对未知杂质的结构进行了分析,其与盐酸普萘洛尔的一级质谱和二级质谱见图5。通过LC-MS 研究发现,该未知杂质不仅与盐酸普萘洛尔有相近的保留时间,其一级质谱和二级质谱也非常接近,仅二级质谱的碎片离子丰度有一定差异,故推测其应为盐酸普萘洛尔的同分异构体,且结构相似。
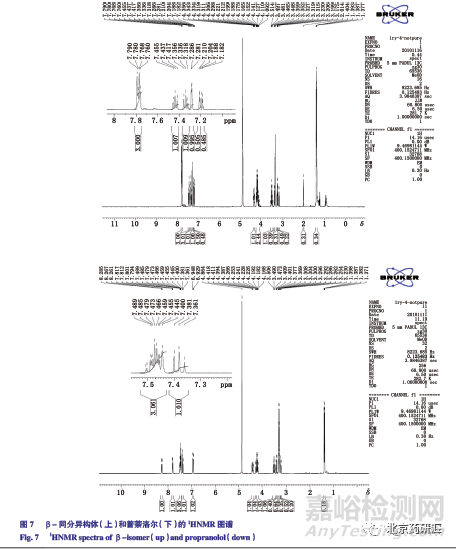
结合紫外光谱信息和盐酸普萘洛尔的合成工艺初步推测未知杂志结构为侧链接在萘环β 位所形成异构体见图6,该杂质来自合成原料α- 萘酚中混入的β- 萘酚的反应产物。
依据上述推断,进一步以β- 萘酚为底物合成了β- 同分异构体,其在LC-MS 上的保留时间和一级质谱、二级质谱信息上与未知有关物质完全一致。进一步通过质谱、核磁共振等方法对所合成的β-同分异构体进行了结构确证,所有结果与推测完全一致见图7。依据盐酸普萘洛尔的结构和合成工艺分析,正常的合成和贮存过程产生β- 位的异构体可能性微乎其微,合成底物α- 萘酚(盐酸普萘洛尔的合成原料)中混有的β- 萘酚应为产生β- 异构体主要来源。
1.4.2 毒性预测
查阅相关资料,目前尚缺乏对未知杂质β- 同分异构体药理毒理研究资料,因此本研究使用Simulations Plus 公司的ADMET Predictor计算软件对该杂质进行了毒性预测。预测结果显示该杂质口服急性毒性低,表现出一定的对梨形四膜虫毒性,鱼类毒性、蜜蜂毒性,无染色体变异性,不会产生致突变性,不会致癌,大鼠急性毒性低,无其他明显毒性,在体内不可降解。预测结果表明,该杂质的毒性均较小,但仍需进一步进行药理毒理性质研究,该杂质的存在具有一定的潜在风险,应予以控制。
2.6 方法学验证
2.6.1 耐用性试验
取上述SST 溶液,色谱条件其他参数不变,分别考察该方法在不同柱温(27、30、35 ℃)、不同流速(1.1、1.2、1.3 mL·min -1 )、不同批次CERI L-Column 3(250 mm×4.6 mm,5 μm)色谱柱的耐用性。
不同柱温的影响:由表7 可知,温度在27 ~35 ℃范围内变化,色谱峰特性略有变化。温度升高保留时间迁移,主峰与相邻杂质峰分离度和理论板数也有所升高,杂质B 相邻双峰分离度略有下降,拖尾因子略有降低,综合评判柱温设置为30 ℃,主峰与相邻杂质峰分离度满足基线分离。
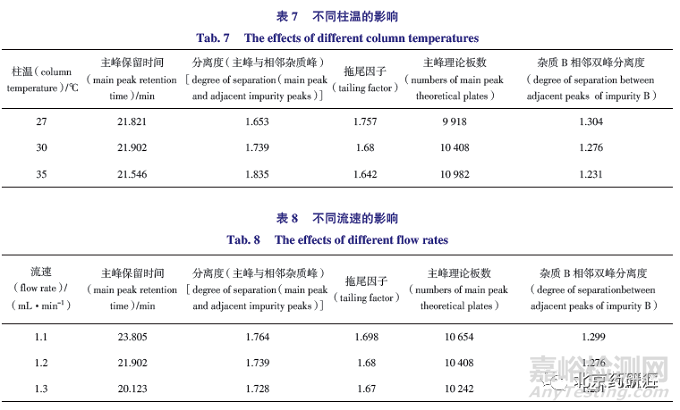
不同流速的影响:由表8可知,流速在1.1 ~1.3 mL·min - 1 范围内变化,色谱峰特性略有改变,随着流速增加,主峰保留时间缩短,主峰与相邻杂质峰分离度和杂质B 相邻双峰分离度略降低,拖尾因子也随着降低,综合评判采用1.2 mL·min -1 流速。
不同批次色谱柱的影响:由表9 可知,研究采用的CERI 品牌不同批号色谱柱影响不大,均可以将β- 同分异构体与主峰基线分离。
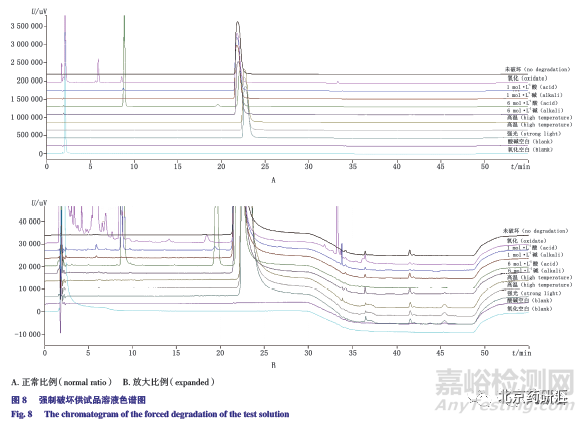
2.6.2 专属性试验
2.6.3 重复性试验
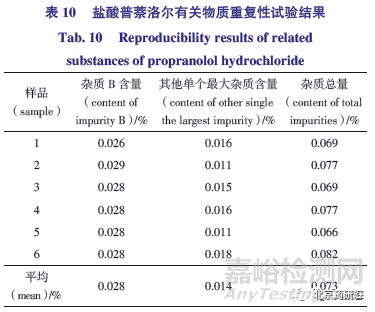
(1)如表9 所示,6 份供试品均检测出杂质B,结果在0.026%~0.029%,其它已知杂质均未检出;6 份样品中其他未知最大杂质含量在0.011%~0.018%,杂质总量均小于0.1%,各杂质含量基本一致,重复性良好,符合有关物质精密度试验要求。
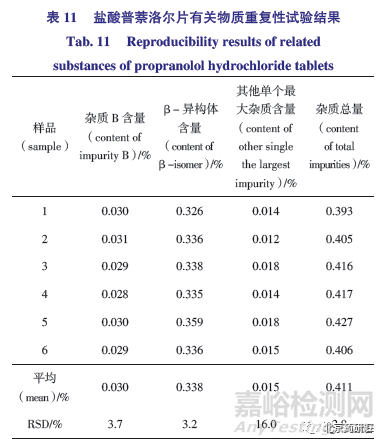
(2)如表11 所示,6 份供试品均检测出杂质B,结果在0.028%~0.031%;均在主峰前发现β- 异构体峰,含量在0.326%~0.359%,其他已知杂质A、B、α- 萘酚均未检出;6 份样品其他单个最大杂质均在0.012%~0.018%。因为该样品中检出β- 异构体,杂质总量在0.393%~0.427%,各杂质含量基本一致,重复性良好,符合有关物质精密度试验要求。
2.6.4 溶液稳定性试验
取上述重复性试验供试品溶液,分别在下述时间点进样分析,记录色谱图,考察杂质峰面积变化情况。
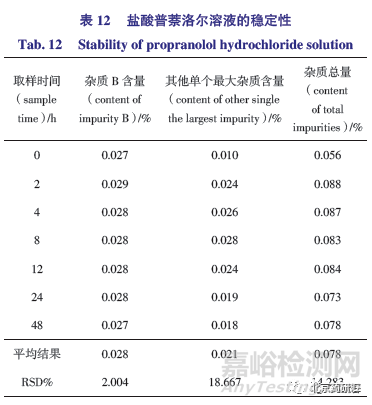
(1)如表12 所示,0~48 h 内不同时间点进样盐酸普萘洛尔原料药供试品溶液均检测出杂质B,含量结果在0.027%~0.029%,其他已知杂质均未检出;其他单个最大杂质含量在0.010%~0.028%,无新的杂质检出,杂质峰面积之和虽略有变化,但是显示和放置时间关系不大,杂质总量未超过0.1%。结果表明,供试品溶液在48 h 内可保持基本稳定。
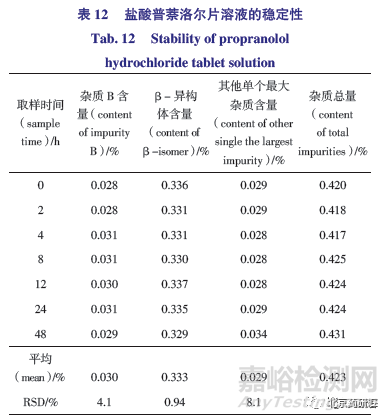
(2)如表12 所示,不同进样时间盐酸普萘洛尔片供试品均检测出杂质B,结果在0.028%~0.031%;均在主峰前发现β- 异构体峰,含量在0.329%~0.337%,其他已知杂质A、C 及α- 萘酚均未检出,供试品溶液在48 h 内可保持稳定,没有新的杂质检出,符合有关物质试验要求。
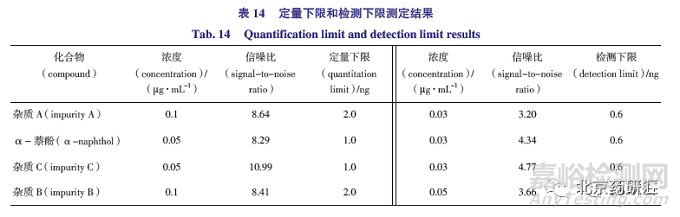
2.6.5 定量下限和检测下限
取盐酸普萘洛尔对照品(USP)和已知杂质对照品各适量,分别用乙腈-水(50∶50)溶解并定量稀释,精密量取20 μL 进样,注入色谱仪,记录色谱图。当信噪比约为10∶1 时测得盐酸普萘洛尔定量限为4 ng(0.2 μg·mL -1 ),相对供试品浓度为0.02%;当信噪比约为3∶1 时测得检测限为2 ng(0.1 μg·mL -1 ),相对供试品浓度为0.01%,检测限浓度约为对照品溶液浓度(1 μg·mL -1 )的1/10,灵敏度可满足测定需求,其他已知杂质的定量限和检测限结果见表13。
2.7 样品检测
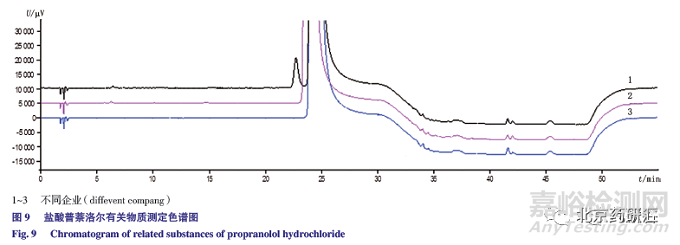
2.7.1 盐酸普萘洛尔原料药
采用拟定方法检测3 家企业提供的3 批样品,结果见表15 和图9,其中企业-2 和企业-3 的单个杂质和杂质总量均在0.1% 以下,而企业-1 由于含有β- 异构体,含量为0.347%,杂质总量为0.389%。
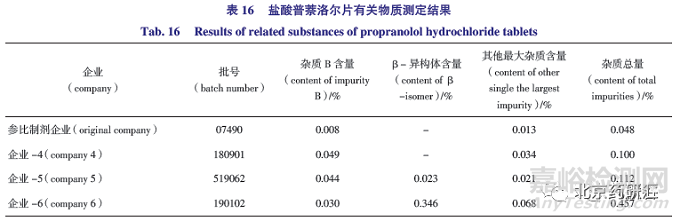
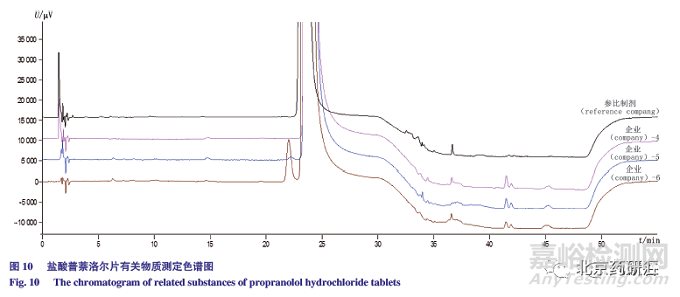
2.7.2 盐酸普萘洛尔片
采用拟定方法检测参比制剂和3 家仿制药企业共4 批样品,结果见表16 和图10,其中参比制剂不含β- 异构体,杂质总量仅为0.048%,仿制药企业4 不含β- 异构体,而仿制药企业5 和6 均检出β- 异构体,含量分别为0.023% 和0.346%,仿制药企业4 和5 单个杂质和杂质总量均低于0.1% 和0.2%,而企业-6 由于含有β- 异构体,杂质总量为0.457%。
3 讨论
3.1 新的有关物质检测方法的建立
目前,已报道测定盐酸普萘洛尔的分析方法有色谱法、毛细管电泳法、电化学分析方法、光纤传感分析技术等。色谱法具有分离效果好、辅料无干扰 、快速、简便等优势,已广泛应用在盐酸普萘洛尔的检测中。各国现行版药典均采用复杂的混合离子对色谱法检查盐酸普萘洛尔有关物质,该方法存在的问题是,实测发现峰形和柱效均不理想,对普萘洛尔的结构类似物(如α- 萘酚、β- 酚异构体)不能有效分离,且292 nm 的检测波长灵敏度十分有限。本探索研究建立了新的HPLC 测定方法,用新型碱性色谱柱系统替代混合离子对方法,并采用更为灵敏的230 nm 波长进行检测。该方法不仅对欧洲药典收载的3 种已知杂质(有关物质A、B、C)可以有效分离,同时对普萘洛尔的结构类似物具有更好的分析能力。并且本方法流动相水相的pH 为11.0,因此需要选择耐碱性比较强的色谱柱。通过筛选比较,CERI 生产的L-column3 C18 色谱柱采用了独特的气态峰尾技术和新开发的PCS 硅胶技术,稳定性较好,适用的pH范围为1~12,具有较高的耐碱性。研究也证实该色谱柱的不同批次均表现较好的稳定性和耐受性,因此推荐操作中采用该色谱柱或效能相当的色谱柱。
经对不同生产企业原料药和片剂样品检测以及同批次样品在不同的保存条件下的样品检测结果进行分析,各特定杂质、单个最大杂质和杂质总量均无显著性差异,进一步表明本品有关物质主要来自原料药,与制剂生产工艺和保存条件关系不大,风险控制的重点应控制原料药。
3.2 β- 异构体研究
根据新建的有关物质检测方法,发现原料药生产企业-1 含有β- 异构体,而2 家仿制药生产企业5 和6 也检出此杂质。经调研发现,此2 家仿制药生产企业的原料药来源均为原料药生产企业-1,其中仿制药生产企业-6 和原料药生产企业-1 为同1 批次,2 者β- 异构体含量相似,表明该杂质在制剂加工中未有变化,而仿制药生产企业-5 原料药来源为不同批次,β- 异构体含量较低,表明该原料药生产企业合成工艺不稳定,导致各批原料间β- 异构体含量不一致。依据盐酸普萘洛尔的结构和合成工艺分析,正常的合成和贮存过程产生β- 异构体可能性微乎其微,在强制破坏性试验中也未产生β- 异构体,因此推测合成底物α- 萘酚(盐酸普萘洛尔的合成原料)中混有的β- 萘酚应为产生β- 异构体主要来源。由于目前尚缺乏对β- 异构体药理毒理研究资料,故应对β- 构异构体的潜在风险予以关注,并设法控制其限量,进一步研究β- 异构体性质,制定更为科学的限度。根据新建的检测方法,发现参比制剂检测并无此杂质,而国内中检院提供对照品以及部分样品均含有此杂质,应在对照品制备和仿制药评价中予以关注。此外,考虑到β- 萘酚的反应位阻较低,类似的情况在其他使用α 酚萘酚作为合成底物的药物中也可能存在,应同样加以重视,同时对作为原料药合成底物的α- 萘酚应制定更为严格标准,从源头上控制产生此类杂质的潜在风险。
4 结论
本研究对盐酸普萘洛尔原料药和片剂现行标准中的有关物质测定方法进行了考察及优化,在此基础上建立的新方法灵敏度高,专属性强,操作简便,能将游离萘酚检查纳入有关物质检测中,并有效检出现行标准未能检出的β- 异构体,为《中国药典》2020 年版中盐酸普萘洛尔质量标准的修订提供了参考。综合结构和合成工艺分析,推测该物质为盐酸普萘洛尔的β- 异构体,是盐酸普萘洛尔的合成底物α- 萘酚中混有β- 萘酚而产生的合成副产物,由于该β-异构体药理毒理性质尚无相关详细资料,该杂质的存在具有一定的潜在风险。通过研究,还确认了盐酸普萘洛尔高风险杂质及质量控制关键点,指导企业优化相应的生产工艺,提升产品质量,为科学监管提供技术保障和执法依据,促进国产仿制药质量的不断提高,为一致性评价工作提供有力的技术支撑。

来源:Internet


