您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-02-16 22:06
摘要: 目的 研究FDA《特定药物的生物等效性指导原则》对透皮贴剂生物等效性研究的相关规定,为我国仿制药质量和疗效 一致性评价工作提供借鉴和帮助。方法 根据美国FDA2018年4月发布的部分透皮贴剂目录,结合FDA特定药物的生物等效性指导原则进行分析,从具体研究类型入手,着重对部分品种从试验设计、试验方案、受试者选择及纳入与排除标准、检测物质、生物等效性豁免等方面进行介绍分析。结果 与常规剂型相比,FDA《特定药物的生物等效性指导原则》规定透皮贴剂 仿制药生物等效性研究,除了开展以药动学为终点的生物等效性研究外,还应遵循FDA《ANDA: 评价经皮和局部给药系统的黏附力( 草案) 》及《ANDA: 评价经皮和局部给药系统的刺激性和致敏可能性( 草案) 》的整体思路,开展黏附性研究、皮肤刺激 性和致敏性研究,并从多个方面进行较为详细的规范描述,对透皮贴剂仿制药的发展具有重要的推动作用。结论 在国家药品监督管理局尚未颁布针对透皮贴剂生物等效性研究相关规定的背景下,《特定药物的生物等效性指导原则》对透皮贴剂相 关规范对我国正在进行的仿制药质量和疗效一致性评价工作具有一定指导和借鉴意义。
透皮贴剂属于经皮给药制剂,是一种用于完整 皮肤表面,能将药物输送透过皮肤进入血液循环系 统发挥全身作用的剂型[1-2]。经皮给药制剂分为传 统外用制剂和现代经皮吸收制剂两大类。传统外用制剂也称为皮肤局部给药系统,主要作用于皮肤局 部或者局部皮肤下的各层组织,常见制剂种类包括 软膏剂、乳膏剂、糊剂、凝胶剂、喷雾剂、涂剂、气雾 剂、巴布剂、泡沫剂和散剂等。现代经皮吸收制剂一 般主要是指透皮贴剂。虽然透皮贴剂存在药物吸收慢,以及不适用于对皮肤有强刺激性和过敏性药物等的局限性,但与常规注射剂和口服制剂等相比,透皮贴剂仍具有多项优点: 一是给药方便,适用于低龄儿童或吞咽困难患者,患者顺应性高; 二是避免肝脏 首过效应及药物在胃肠道的降解,避免胃肠道因素 对药物吸收产生的影响; 三是能够长时间持续释放药物,避免口服给药引起的血药浓度的峰谷现 象[3-4]。透皮给药概念由 Stoughton于1965年首次 提出,1979年,美国ALZA公司开发的第1个透皮贴剂产品东莨菪碱贴剂在美国获批上市,经过40余年 的发展,透皮贴剂目前已成为第3代药物制剂的研究热点之一[5],其主要用于各种长期性和慢性疾 病,包括心血管疾病、精神病、过敏性疾病以及长期 性胃肠疾病等。
生物等效性( bioequivalence,BE) 是我国正在进行的仿制药质量与疗效一致性评价工作的重点之 一,也是目前公认的衡量仿制药品质量和疗效的有 效手段,目的是证明仿制药品和参比制剂生物等效, 以桥接与参比制剂相关的临床前试验和临床试验[6-8]。截至2021年3月,我国药监机构已经公布 的与透皮贴剂相关的生物等效性最新官方技术指导 原则包括: 《化学药物制剂人体生物利用度和生物等效性研究技术指导原则》( 2007年8月颁布) 《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》( 2015年11月颁布) 《人体生物等效性试验豁免指导原则》( 2016年5月颁布) 《生物等效性研究的统计学指导原则》 ( 2018年10月颁布) 。国外先进监管机构( 如EMA和FDA) 已陆续公布了透皮贴剂相关技术要求或指南。《中华人民共和国药典》2020年版第4部及国家药品管理局药品审评中心2020年12月发布的《化学仿制药透皮贴剂药学研究技术指导原则( 试行) 》对透皮贴剂的黏附力、含量均匀度、重量差异及释放度等药学质量检测方法提出了相关要求; 国家药品管理局药品审评中心2021年3月发布的《皮肤外用化学仿制药研究技术指导原则( 试行) 》结合皮肤外用化学仿制药的制剂特点,对皮肤外用化学仿制药在开发过程中的药学研究、非临床研究提出 了具体指导,并对局部皮肤外用仿制药的生物等效性研究提出技术要求,但是尚未对透皮贴剂的生物等效性研究提供具体指导。
虽然FDA在《以药动学为终点评价指标的仿制药生物等效性研究指导原则( 草案) 》中对仿制药BE研究的思路清晰、具体,但是关于透皮贴剂的BE研究方案仍然不够明确[6]。作为FDA《以药动学为终点评价指标的仿制药生物等效性研究指导原则( 草案) 》的补充,FDA于2010年开始,陆续公布针对特定药物的生物等效性指导原则,并持续更新药物品种和内容,形成特定药物指导原则数据库数据库。截至2021年2月9日,FDA在其网站中已公布了1866个特定品种的生物等效性指导意见,其中包括 透皮贴剂特定药物的生物等效性指导原则[9]。
鉴于近年来,透皮贴剂仿制热度骤增,在我国尚未发布针对透皮贴剂的生物等效性指导原则的背景 下,本研究结合中国药学会承担国家药品监督管理局有关翻译美国FDA《特定药物的生物等效性指导 原则》相关工作,对FDA对透皮贴剂生物等效性有 关内容进行介绍和分析,为我国透皮贴剂仿制药的研发和监管提供参考。
一、方 法
本研究将根据2018年4月23日美国FDA发 布的部分透皮贴剂名单结合FDA特定药物的生物等效性指导原则进行分析,针对以药动学为终点的生物等效性研究、黏附性研究、皮肤刺激性和致敏性研究等3个具体研究类型,着重对部分品种从试验设计、试验方案、受试者选择、检测物质及生物等效 性豁免等方面进行介绍分析。
二、 结 果
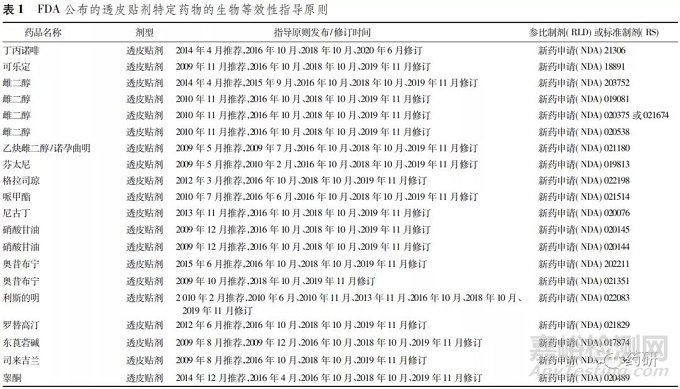
2. 1 美国FDA发布的透皮贴剂名单
2018年4月23日美国FDA发布了《FYs 2013- 2017 GDUFA Science and Research Report: Transder- mal Drug Products》,公布了包括18个透皮贴剂的名 单目录[10]。结合《特定药物的生物等效性指导原 则》,对相关品种进行了梳理( 表 1) 。《特定药物的生物等效性指导原则》对具体品种透皮贴剂生物等 效性从以药动学为终点的生物等效性研究、黏附性研究、皮肤刺激性和致敏性研究等3个方面作了较 为详细说明。
2. 2 具有以药动学为终点的生物等效性研究
2. 2. 1 具有以药动学为终点的生物等效性研究试验设计 美国FDA在2013年颁布的《以药动学为终点评价指标的仿制药生物等效性研究指导原则》 ( 草案) 中,明确提出了根据药物特点,可选用3种不同的设计。一是双制剂、双序列、双周期、单剂量、交 叉试验设计: 对于一般药物,推荐选用此试验设计, 纳入健康志愿者参与研究。在这种设计中,每位受试者依照随机顺序服用受试制剂和参比制剂。二是单 剂量、平行试验设计: 对于半衰期较长( 大于 24h) 的药物,可选择该试验设计,即每个制剂分别在具有相似人口学特征的两组受试者中进行试验。三是重复 序列设计: 该试验设计是前2种的备选方案,重复序列设计是指将同一制剂重复给予同一受试者,可设计为部分重复( 单制剂重复,即三交叉) 或完全重复( 两制剂均重复,即四交叉) 。重复序列设计在药物变异性较高的情况下很有用,优势在于纳入较少的受试者进行试验,即可获得相似的统计效力。其中绝大部分药品推荐第1或第2种试验设计。
针对透皮贴剂的以药动学为终点的生物等效性研究试验,FDA《特定药物的生物等效性指导原则》 指出申请人应遵循FDA目前在指南中的思路在ANDA下提交药物以药动学终点进行生物等效性研究用于设计和进行药动学生物等效性研究。该指导原则推荐的试验设计类型主要为单剂量,双治疗,双周期交叉体内试验。申请人应随机抽取受试者,在指定的研究期间接受受试制剂或参比制剂。在可能的情况下,在第2次研究期间,敷用 TDS 部位应与第1次研究期间部位相同,但在身体对侧。该指导原则着重指出TDS与皮肤的接触对TDS在体内性能至关重要,当TDS失去对皮肤的黏附时,其药动学可能发生改变。因此,在整个药动学研究过程中,应监测并记录每个TDS黏附情况。申请人应提前 规定其药动学终点统计分析的纳入标准,并对符合方案人群进行基本药动学分析,然而,无论TDS黏附评分如何,无论药动学终点的统计分析纳入标准 如何,都应在所有采样时间内收集和分析所有受试者的药动学样本。研究方案中应规定在整个研究过程中避免发生包括将TDS脱落区重新贴回皮肤及向TDS施加压力或增强TDS与皮肤的黏附力( 例如覆盖物) 等刻意行为,以确保数据可靠。
2. 2. 2 给药剂量 FDA该指导原则对具体品规的给药剂量给予了具体规定( 表2) 。同时规定,除非 另有说明,TDS应该应用于所有受试者的相同解剖部位,从参比制剂批准标签中推荐的剂量中选择剂 量,并贴敷7d。
2. 2. 3 受试者选择 FDA对生物等效性试验受试者的选择一般应符合以下要求: 年龄在18岁以上 ( 含18岁) ; 应涵盖一般人群的特征,包括年龄、性别和种族; 如果药物拟用于两种性别,那么研究入选的男性和女性应占相似的比例; 如果药物主要适用于老年人群,那么入选受试者应尽可能多地选择老年人( 60岁以上的人) ; 入选受试者的例数应足以使生物等效性评价具有足够的统计学效力,但并不 要求所划分的亚组也满足统计学要求( 一般不鼓励划分亚组进行统计分析) [6]。《特定药物的生物等效性指导原则》对大部分透皮贴剂受试者做了进一步的规定,规定受试者为男性和非怀孕女性,非哺乳女性,普通人群。此外,还对部分药品做出进一步的规定: 如雌二醇的受试者为没有雌激素治疗禁忌的绝经后妇女; 奥昔布宁的受试者为非怀孕女性和非哺乳女性; 乙炔雌二醇的受试者为健康非怀孕、非哺乳期的女性,激素类避孕候选人; 尼古丁的受试者为吸烟者,包括男性和非怀孕女性,未哺乳女性; 睾酮的受试者为为睾酮缺乏( 性腺机能减退型) 的男性,其他方面健康,年龄在18 ~ 65岁之间。
上述透皮贴剂受试者选择方面,FDA建议申请人可附加受试者纳入标准和排除标准。例如雌二醇受试者纳入标准: ①不吸烟,绝经后女性受试者,无雌激素治疗禁忌。“绝经后期”定义为自发性闭经12个月或自发性闭经6个月,血清促卵泡激素水平 为> 40 mu·mL-1或术后6周双侧卵巢切除包括或不包括子宫切除。②基线收缩压不大于140mm Hg,舒张压不大于80mmHg。③受试者> 40岁,在参加研究之前有乳房 X 光检查阴性的记录( 在筛查 时或参加研究后9个月内获得) 和正常的临床乳房检查。完整子宫的受试者进行基线阴道超声检查, 发现子宫内膜不活跃,内膜厚度小于4mm。雌二醇排除标准:1.男性受试者。2.绝经前期,围绝经期, 怀孕或哺乳期的受试者。3.未确诊的异常生殖器出血。4.已知、疑似或有乳腺癌病史。5.已知或疑似雌激素依赖性肿瘤。6.有子宫内膜癌的病史或存在子宫内膜癌的风险因素。7.吸烟或体重 > 90kg的受试者。8.患有活动性深静脉血栓,肺栓塞,或这些情况的病史。9.静脉血栓或动脉血栓的高风险。10.活动性动脉血栓性疾病( 中风和心肌梗死等) ,或有此类病史。11.与参比制剂发生过敏反应或血管水肿。12.肝脏损伤或疾病。13.蛋白C,蛋白S,或抗凝 血酶缺乏,或其他嗜血性疾病。14.胆汁淤积性黄疸、 高血压、冠心病或其他严重心脏病、糖尿病、高胆固醇血症、高钙血症、甲状旁腺机能减退、高甘油三酯血症、系统性红斑狼疮、肾功能损害、子宫切除术后 残余子宫内膜异位症、哮喘、癫痫、偏头痛、卟啉症或肝血管瘤史。15.麻醉品滥用、药物滥用或酗酒史。16.在给药前6个月内,使用雌激素丸治疗或黄体酮注射药物治疗。17.给药前3个月内,黄体酮植入剂和单独雌激素注射药物治疗。18.在给药前8周内,口服雌激素和/或口服或宫内黄体酮治疗。19.在给药前4周内,单独使用透皮雌激素或透皮雌激素/黄 体酮产品。在给药前1周内,使用阴道激素产品 ( 套圈、乳剂,凝胶剂) 。20.手术前4~6周内与血栓栓塞风险增加或长期静养期间相关的手术类型。20服用甲状腺激素替代疗法。21.服用圣约翰草、抗惊 厥药、保泰松、利福平、利福布汀、奈韦拉平或依非韦伦等CYP3A4诱导剂。22.服用CYP3A4抑制剂红霉 素、克拉霉素、酮康唑、伊曲康唑、利托那韦、奈非那韦或葡萄柚汁等。其他透皮贴剂受试者纳入和排除标准有不同调整。此外,睾酮的在受试者纳入标准 方面,FDA要求受试者为睾酮缺乏( 性腺机能减退 型) 的男性,年龄在18 ~ 65岁之间。
2. 2. 4 生物等效性试验豁免 FDA《特定药物的生物等效性指导原则》涉及生物等效性试验豁免的 情况主要包括3种: ①药物剂型相同,但规格不同, 其活性成分和非活性成分的比例( 处方) 与已进行过BE试验的规格相似的情况下,根据溶出试验和最大规格BE试验可以申请1个或多个较小规格的BE试验。②在FDA颁布的2017年版《基于生物药剂学分类系统的口服固体速释制剂BA/BE豁免原则》与2010版相比,增加了BCSⅢ类的BA/BE豁免及增加了NDA阶段的BE豁免要求,并明确规定BCS I或III类药物与其他BCS分类药物组成的组合物不适用于BE豁免。③一些属于药效研究实施方案( drug efficacy study implementation,DESI) 的有效药物,尚未发现已知或疑似的生物等效性问题, FDA不推荐进行生物等效性试验。
与口服固体制剂的规格即载药量不同,由于透皮贴剂中药物的含量通常高于使用过程中的递送剂量以达到临床有效给药率,且给药后贴剂中存在药物残 留,因此,采用透皮贴剂的载药量( 所含药物总量) 标示其规格是不合理的,FDA对透皮贴剂的规格通常是用其表观释药速率表示,即递送量/释放时间( 例如,**mg·d-1或**mg·h-1或**mg·24 h-1 ) 表示。不过,目前国内已上市透皮贴剂的规格包括载药量、载药量/贴剂面积、递送速率等多种表达方式。FDA《特定药物的生物等效性指导原则》中提出制剂处方相似的基于高规格可申请豁免其低规格的透皮贴剂为丁丙诺啡、可乐定、雌二醇、哌甲酯、尼古丁、罗替高汀及睾酮等; 明确提出以下透皮贴剂不适合申请豁免,包括乙炔雌二醇/诺孕曲明、格拉司琼、奥昔布 宁及东莨菪碱等。此外,基于规格为25 mg·h-1芬太 尼可接受的生物等效性研究,可考虑豁免规格为12. 5、37. 5、50、75和100 μg·h-1的体内试验; 基于规格为0. 4 mg·h-1硝酸甘油可接受的生物等效性研究,可考虑豁免规格为 0. 1、0. 2、0. 3 、0. 6 和0. 8 mg· h-1的体内试验; 基于规格为9. 5 mg·24 h-1利斯的明可接受的生物等效性研究,可考虑豁免规格为4. 6 和13. 3 mg·24 h-1的体内试验; 基于规格为6mg· 24h-1司来吉兰可接受的生物等效性研究,可考虑豁免规格为9和12mg·24 h-1的体内试验。不同规格的透皮贴剂制剂处方相似标准包括: ①受试制剂每单位活性表面积中活性成分和非活性成分的数量相同。②申请豁免的受试制剂每个规格的活性表面积与已进行过BE试验规格的比值与参比制剂每个对应剂量活性表面积与已进行过BE试验规格参比制剂的比值相同。此外,FDA规定申请豁免透皮贴剂的所有规格体外溶出度试验可接受。
2. 2. 5 参比制剂选择 参比制剂选择是生物等效性试验的关键点之一。为了避免由于参比制剂使用的不同而可能导致的各仿制品之间发生显著的差异,FDA对于参比制剂的选择很明确,在橙皮书的处方药及非处方药目录中,FDA规定了参比制剂目录。在参比目录中对每个品种的参比制剂都提供了明确信息,并且提供了特定药厂和特定规格等信息[11]。因此,在FDA《特定药物的生物等效性指导原则》中仅列出参比制剂RLS /RS号,对其他具体信 息未做重复描述。
2. 2. 6 检测物质 FDA一般推荐测定原形药物, 而不是代谢产物。原因是代谢产物的药时曲线主要反映代谢物的生成、分布和消除,而原形药物的药时 曲线比代谢产物的能更灵敏地反映制剂的变化[4]。FDA 同时规定,如果由从原形药物产生的主要代谢产物满足以下两点,则可作为生物等效性试验的检 测物质: 代谢产物基本上产生于进入机体循环系统以前,如首过效应、肠壁细胞内、肠道内代谢等。代谢产物显著影响药物的安全性和有效性。FDA《特定药物的生物等效性指导原则》要求检测的原形、代谢物和其他非原形药检测物质的部分透皮贴剂见表 2。


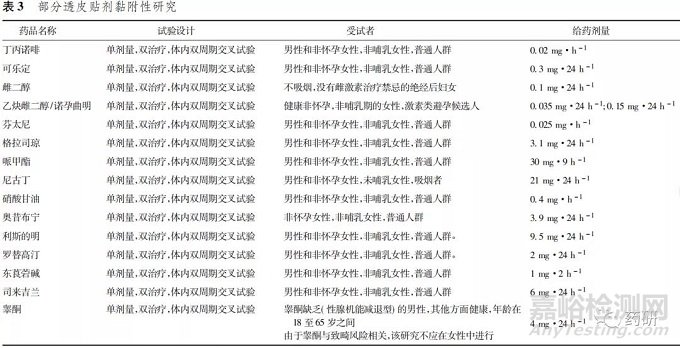
2. 3 黏附性研究
透皮贴剂中药物输送到患者皮肤中或通过患者皮肤输送的药物量部分取决于贴片面积,当透皮贴剂在用药过程中失去黏附力时,可能会减少输送给患者的药物量,影响药物吸收及产品的有效性和安全性。为全面评价其等效性,FDA 建议对仿制产品的黏附力进行评价。FDA《特定药物的生物等效性 指导原则》推荐遵循FDA《ANDA: 评价经皮和局部 给药系统的黏附力指南( 草案) 》中的思路,申请人可选择单独开展黏附性研究,也可与以药动学为终点的生物等效性研究结合合并开展。
FDA《特定药物的生物等效性指导原则》黏附性研究对受试者选择及试验研究设计类型与以药动学为终点的生物等效性研究基本相同( 表3) 。FDA 《ANDA: 评价经皮和局部给药系统的黏附力》指南草案对黏附性研究设计和实施、统计分析和综合评价等方面提供了指导,特别是提供了黏附力采样时间点、附着力评分系统( 按 5 点制评分标准进行评 估) 及黏附力统计等具体方法。附着力评分系统评分标准: 0分: ≥90% 附着( 基本没有翘离皮肤) ; 1分: ≥75%至< 90%附着( 仅有一些边缘翘离皮 肤) ; 2分: ≥50%至<75%附着( 不多于一半翘离皮肤) ; 3分: <50%附着,但没有脱落( 多于一半翘离皮肤,但没有脱落) ; 4分: 贴片脱落( 贴片完全脱离皮肤) [12]。
2. 4 皮肤刺激性和致敏性研究
FDA建议透皮贴剂应考虑进行皮肤刺激性试验和过敏性试验。刺激性是指非口服给药制剂给药后对给药部位产生的可逆性炎症反应,若给药部位产生了不可逆性的组织损伤则称为腐蚀性。过敏性又称超敏反应,指机体受同一抗原再刺激后产生的一种表现为组织损伤或生理功能紊乱的特异性免疫反应,是异常或病理性免疫反应。
2. 4. 1 试验设计及受试者选择 与以药动学为终点的生物等效性研究及黏附性研究的试验设计不同,FDA《特定药物的生物等效性指导原则》 推荐透皮贴剂的皮肤刺激性和致敏性研究试验设计方法为随机、评估盲法、受试者体内重复试验( 表4) 。在受试者选择方面,皮肤刺激性和致敏性研究与前述生物等效性研究及黏附性研究要求相同,不再赘述。
2. 4. 2 给药剂量 FDA对上述透皮贴剂进行皮 肤刺激性和致敏性研究的推荐剂量主要包括3类 ( 表4) : ①给药剂量与生物等效性研究及黏附性研究给药剂量相同,包括乙炔雌二醇 /诺孕曲明、 格拉司琼、奥昔布宁、利斯的明及司来吉兰等。② 给药剂量与生物等效性研究及黏附性研究给药剂量不同,多数为小于生物等效性研究及黏附性研究给药剂量,主要包括可乐定、雌二醇、哌甲酯、尼 古丁、硝酸甘油、罗替高汀碱及睾酮等。③由于安全考虑,本研究不应使用含有活性药物成分的TDS,仅使用TDS载体和阳性对照即可,主要包括丁丙诺啡 、芬太尼及东莨菪碱等。


FDA《特定药物的生物等效性指导原则》指出开展透皮贴剂的皮肤刺激性和致敏性研究,申请人应遵循FDA《ANDA: 评价经皮和局部给药系统的刺激性和致敏可能性指南( 草案) 》的思路进行试验的设计、实施及统计分析等。《ANDA: 评价经皮和局部给药系统的刺激性和致敏可能性( 草案) 》着重推 荐了皮肤反应的两种评分系统: 一是根据皮肤外观的反应评分系统: 未见刺激性为0分; 刚刚可以观察到轻微红斑为1分; 明显的红斑( 肉眼可见的轻微水肿或轻微丘疹反应) 为2分; 红斑和丘疹为3分; 明显的水肿为4分; 红斑、水肿和丘疹为5分; 水泡为6分; 强烈反应( 蔓延到超出皮肤贴敷区域) 为7分。二是其他可观察到的反应评分系统: 皮肤表面轻微发亮为A( 0) ; 皮肤表面明显发亮为B( 1) ; 皮 肤表面发亮并伴有脱皮和皲裂为C( 2) ; F: 皮肤表面发亮并伴有明显裂缝为 F( 3) ; 整个或部分贴敷部位被覆一层渗出物干膜为G( 3) ; 出现小斑点性糜烂和/或疮痂为H( 3) [13]。
三、 结 语
与普通口服固体制剂相比,透皮贴剂是通过皮肤吸收药物进入人体血液循环并达到有效血药浓度以实现疾病治疗或预防,存在生物等效性研究复杂、 标示规格不统一等问题。鉴于国内尚无专门的透皮贴剂生物等效性研究指导原则,建议相关部门参考FDA生物等效性指导相关内容开展研究。

来源:Internet


