您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-08-21 21:19
监管科学发展下药品审批提效的实践探讨
Practices of Improving Efficiency in Drug Approval Under the Regulatory Science
摘 要 Abstract
药品监管科学研究在最近10 年取得了长足的发展。不管是从研究论文的产出,还是监管机构的法规以及指南文件的及时发布,均体现了顺应当前科技发展的需求。面对持续增长的审评压力,监管机构在资源有限的情况下,通过设置监管促进路径、开启共享审评模式等方式,提高审评效率,加速新药上市。展望未来的药品审评审批发展,将始终坚持以患者为中心的宗旨,全面考虑和平衡多个利益相关方的诉求,从而真正加速并优化创新产品从法规审评到患者可及的全流程。
There is significant progress in the research of drug regulatory science in the last ten years. Both the output of research articles and the timely publication of regulations and guidance reflect the demands of adaption to current rapid development of science and technologies in pharmaceutical industry.With increasing review pressure and limited resources, global regulatory agencies have leveraged facilitated regulatory pathways and shared review mode to improve the review efficiency and accelerate the approval of new drugs. Drug regulatory review in the future should adhere to the principle of patient-centricity, comprehensively balance the demands from multiple stakeholders, so as to optimize and truly accelerate the whole process of innovative drugs from regulatory review to patient affordable access.
关键词 Key words
监管审评;监管科学;共享审评模式;监管促进路径;卫生技术评估
regulatory review; regulatory science; shared review mode; facilitated regulatory pathways; health technology assessment
进入21 世纪第三个十年,科学技术进步、研发生产率提高,加上2020 年新冠肺炎疫情暴发,使得全球医药及医疗用品需求不断增长,促使整个生物医药产业发展快速推进。活跃的生物技术开发,对生命科学的积极探索,推动了创新疗法层出不穷。如今的先进疗法,如细胞治疗和基因治疗,给标准治疗提供了更多选择。从个性化治疗方案,到超个性化疗法,再到一次性疗法,这些新技术对于现有监管法规和技术审评都带来了巨大挑战。不论是从政府职责、行业呼声还是社会民意,都清楚地传递了同样需求:监管法规需要跟上创新的步伐。
令人兴奋的是,近几年来,不论是国外监管机构还是我国药品监管部门,都能够与时俱进,及时发布多项指导原则和法规文件来规范和引导这些创新疗法的研发。比如国家药品监督管理局药品审评中心在2021 年4 月发布的《用于产生真实世界证据的真实世界数据指导原则(试行)》,在业界引起了巨大反响。值此我国药品监管科学行动计划2 周年之际,本文探讨了药品审评审批的现状、挑战与应对。
01近年全球药品监管科学研究发展迅速
全球监管科学经过10 年发展,取得了长足的进步,并收获了大量研究成果。根据科睿唯安Web of ScienceTM 核心合集的统计,2011~2020 年,全球监管科学研究论文发表超过15 000 篇,其中美国发表了6359 篇、英国2175 篇、德国1289 篇、法国884 篇、日本493 篇,我国学者在近几年也快速跟上,共计发表585 篇。
从每年的论文发表量来看,全球主要国家和地区监管科学研究论文发表均呈现持续增长趋势。2020 年,全球监管科学研究论文发表超过2000 篇,其中美国产出777 篇。我国也从2011 年的17 篇,增加到了2020 年的106篇(图1)。
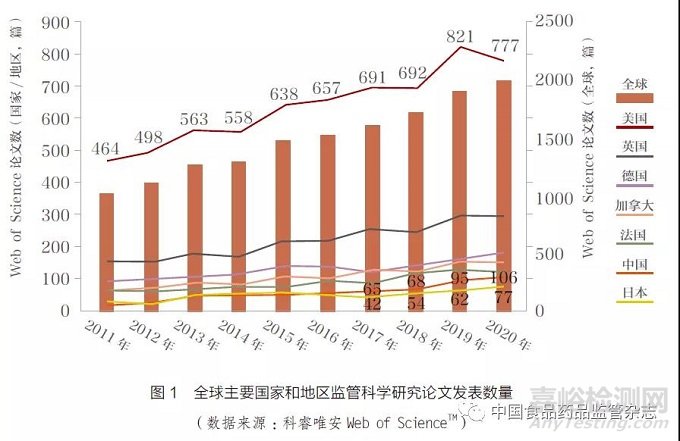
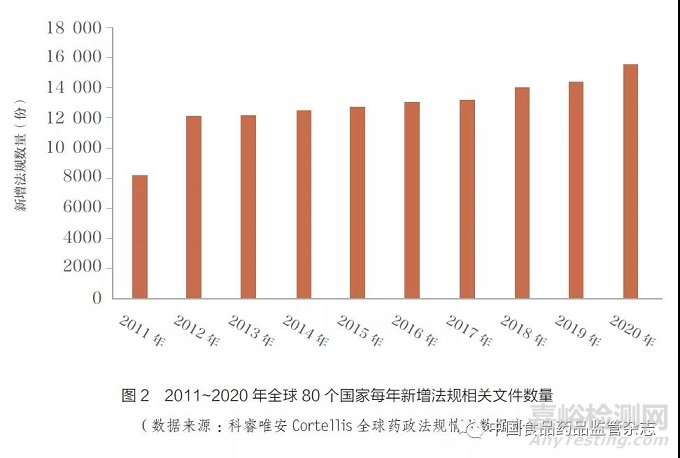
除了全球监管科学研究论文发表外,全球监管法规和指南数量也呈现明显增长。根据科睿唯安Cortellis 全球药政法规情报数据库(Cortellis Regulatory IntelligenceTM)统计,2020 年全球80 个主要药政法规国家新增法规相关文件数量达到了15 520 份(图2)。
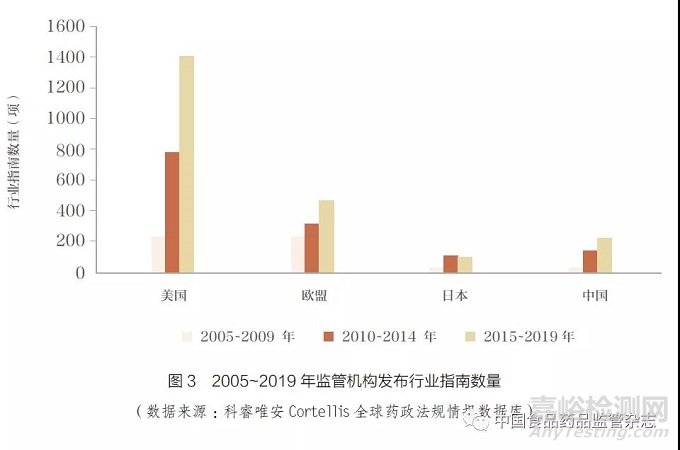
与此同时,监管机构发布的行业指南数量在2005~2019 年也呈现飞速增长。2015~2019年,美国发布的行业指南超过了1400 项,中国也达到了230 多项(图3)。
对于监管机构而言,根据行业发展变化,能够快速响应并发布相应的法规文件加以规范和引导,体现了监管机构的职责和担当。对于企业尤其是面向全球市场的跨国企业而言,同步更新、研究学习如此多数量、快速变化的法规文件和行业指南,亦是一个艰巨的挑战。同样,我国立志于国际化的优秀企业也需要积极、前瞻性地思考如何系统性面对全球纷繁复杂、差异化的监管法规要求。
02监管机构审评压力不断增加
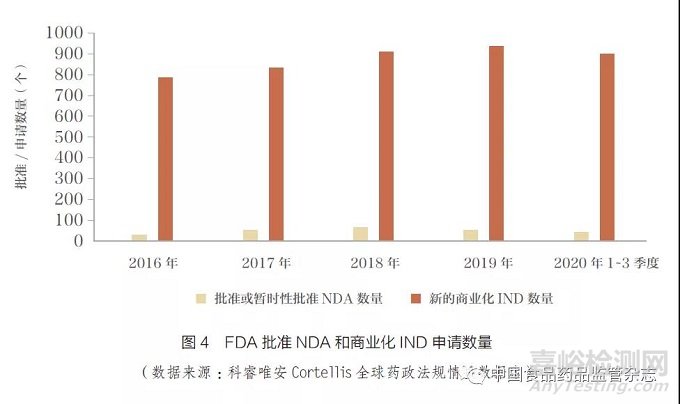
通过分析2016~2020 年美国食品药品监督管理局(FDA)新药上市申请(NDA)和商业化新药临床试验申请(commercial IND) 数量后发现[1], 这5 年FDA 平均每年批准50 个左右新活性实体(NAE),但是受理的商业化IND 申请年均达到900 项左右(图4)。如果粗略计算FDA审评转化率,从商业化IND 申请到NDA 获批, 大约为18 ∶ 1。由此可以看到大量的 IND 申请,对审评资源带来了压力,但是最终的产出维持在低位(年均50 个左右)。合理有效引导商业化IND申请、及时暂停临床价值或创新价值低的项目,将会对节约监管资源、提高审评效率起到积极的促进作用。
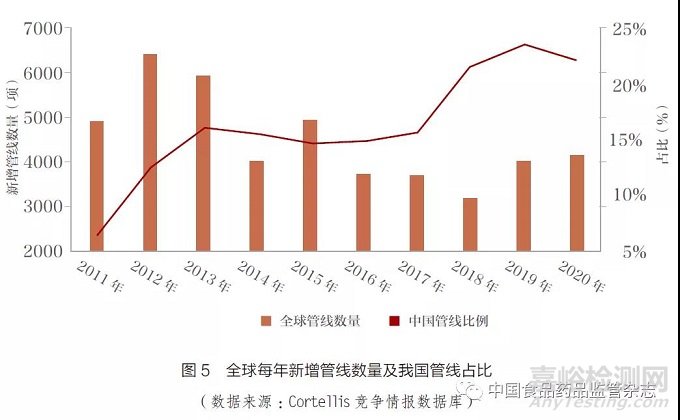
根据Cortellis 竞争情报数据库(Cortellis Competitive IntelligenceTM)统计,近年全球研发管线的新增项目呈现下降趋势,但是技术类型的丰富程度和创新度均有较大提升。对于我国新药研发来说,新药项目在全球管线中的占比大幅增长,从2011 年6% 增加到2020年22%。随着研发项目不断向前推进,可以预见其中大部分项目将在未来几年内,向国家药品监督管理局提交审评申请,国家药品监督管理局药品审评中心的审评压力将会持续加码(图5)。
03监管机构缓解审评压力的实践
为了应对不断增加的审评压力,各国监管机构进行了多方面的尝试,在有限资源情况下提高审评效率,加速新药上市,其中包括设置更多的监管促进路径(facilitated regulatory pathways,FRPs)、开启共享审评模式等。
3.1 共享审评模式
共享审评模式备受关注,有机会成为一种监管机构在区域和跨区域间共享资源、简化与申办方互动的模式。这里探讨2 个案例,ACSS 联盟和Orbis 计划。
ACSS 联盟成立于2017 年,成员包括澳大利亚(A)、加拿大(C)、瑞士(S) 和新加坡(S)[2]。作为共享程序的一部分,各监管机构负责审评不同部分的申请资料。举例来说, 对于尼拉帕尼(niraparib)的审评,加拿大卫生部(HC)完成了临床和质量部分的审评,澳大利亚医疗用品管理局(TGA)完成了非临床审评。对于玻玛西林(abemaciclib),HC 完成了临床审评,TGA 完成了质量和非临床审评。另外,HC 还参考了瑞士医药管理局(Swissmedic)的质量审评报告。虽然当时Swissmedic 未加入该联盟,但在2020 年与HC 和TGA,通过工作共享机制(work-sharing)参与了巴洛沙韦玛波西酯(baloxavir marboxil) 的审评。尽管各监管机构之间共享审评结果,但每家机构均独立决定是否批准新药( 上市许可)。2018~2019 年,HC 和TGA 通过共享审评程序共批准了3 款新化学实体(NCE),分别为尼拉帕尼、阿帕鲁胺和玻玛西林。
Orbis 计划由FDA 发起,针对具有高影响力的肿瘤学产品。这些产品必须具有让患者获益最大的潜力。建立Orbis 计划的一个推动因素是尽早为癌症患者带来高效的治疗方法,尤其是在监管审评周期较长的国家。申请参与的项目通常需要符合FDA 优先审评的标准。目前参与的监管机构有7 家,分别为FDA、TGA、HC、Swissmedic、新加坡卫生科学局(HAS)、巴西卫生监督局(ANVISA)和英国药品和医疗保健产品监管局(MHRA)。
根据统计[3], 在2019 年6月~ 2020 年6 月,通过Orbis计划共收到60 件肿瘤药上市申请(新分子实体申请或添加新适应症的补充申请),分别来自16 个不同项目,最终取得38 个批准。
FDA 和参与机构希望通过Orbis 计划继续进行合作,并有望将该计划扩展到其他国家和更多产品的申请中,包括可能伴随复杂问题的申请(如伴随诊断)。FDA 表示,Orbis 计划已经证明,全球监管合作是可以实现的, 并且可以为癌症患者提供更快获得新疗法的途径。这种合作的共同诉求,可以为治疗方法的开发提供更广泛的信息,尤其是在全球一致应对公共卫生安全的情况下,如新冠肺炎疫情。未来药物开发可能会受益于这种更统一的全球法规监管标准,以最大程度优化重要临床试验设计,达到最快速的市场准入,惠及多国患者。
从这两项共享审评的案例来看,主要是全球龙头企业(年研发投入超过30 亿美元)有能力同时向多国监管机构提交申请。对于立志走向国际市场的我国药企来说,未来可以考虑使用此类注册策略,以达到加快多国市场准入的目的。
3.2 监管促进路径
除了共享审评资源、开启审评互助模式的探索,监管机构提高审评效率的另一个重要举措就是优化监管促进路径的设置,帮助临床急需、解决重大临床需求的创新疗法进入审评快车道。根据科睿唯安年度新药报告显示[4],新分子实体在FDA 从IND 申报到获批平均用时,常规路径为8年,而获得突破性疗法的项目,只需要4.8 年,平均减少40% 时间,这对于申办方来说无疑具有极大的诱惑力。不仅可以大大节约临床开发成本,且争取到了领先对手进入市场的宝贵时间差。
另外,监管促进路径的有利政策对于中小企业来说,意味着更短的开发时间和更小的临床投入,在有限资源的情况下更可能实现从临床试验到上市许可。这一现象也直接体现在跨国药企近几年主导的新药获批占比下降至40% 以下[5]。不仅为创新产品的申办方直接带来市场收益,也保护了创新想法的落地,不至于被实力雄厚的大型企业垄断绝大部分的临床后期开发和全球上市。这对于构建一个良性的创新生态和鼓励创新实施有巨大的保护作用。
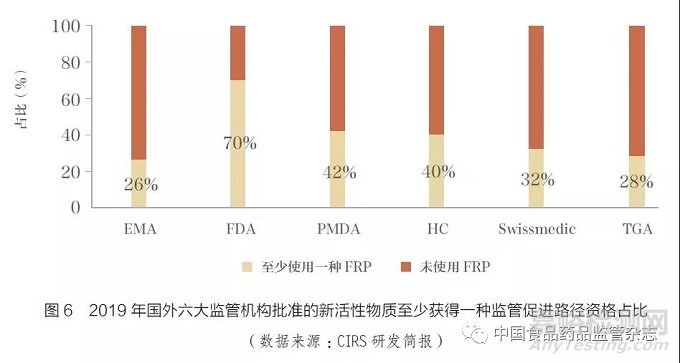
在国外主要监管机构的新药审评审批过程中,监管促进路径的使用已经比较普遍。根据科睿唯安旗下国际监管科学创新中心(CIRS)的研究[3],2019 年监管促进路径在FDA 批准中占比高达70%,中位审评时间缩短了130天左右;欧洲药品管理局(EMA)占比最小,仅为26%,但是审评提速效果明显。2019 年,FDA、EMA 、HC 、Swissmedic、TGA、日本药品及医疗器械综合机构(PMDA)批准的新活性物质至少获得一种监管促进路径资格的占比(图6)。
随着改革开放进程和对药品属性认识的不断深化,监管机构的角色逐渐发生转变,从最初的社会公众健康守护者,到如今兼顾创新医药产品开发的赋能者。监管机构在审评审批过程中通过设置监管促进路径,赋能新药加速获批的成效显著。那么申办方如何看待监管促进路径的价值和实施效果?CIRS 通过一项企业调研[6],尝试解答了这一问题。
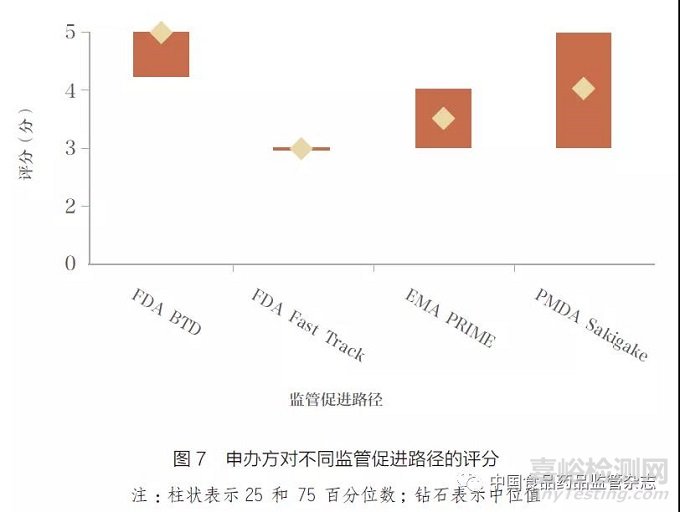
该研究对象包括FDA 突破性疗法(BTD) 和快速通道(Fast Track),欧盟优先药品计划(PRIME) 以及日本先驱疗法(Sakigake)。在调研这4 种监管促进路径的使用情况和申办方的观点后,CIRS 发现:①受访企业普遍认可监管促进路径的积极影响。②绝大多数受访企业认同BTD 的影响最积极,其次为Sakigake、PRIME 与Fast Track( 图7)。③ PRIME 得分较低的原因是, 受访企业认为EMA 没有对所有项目的紧急处理程度保持一致。④受访者认为监管促进路径会对卫生技术评估和支付方造成负面影响或矛盾。⑤创新产品快速的获批并不能保证快速的医保准入,在除美国以外的市场,患者可及性提升不显著。
可以看到,申办方肯定了监管促进路径的价值,并且考虑正在开发的项目会采用单一监管促进路径或单国/ 多国监管促进路径的申请。
对于未来监管促进路径的实践,CIRS 也给出了多项积极的建议,如扩充并优化企业和监管部门的资源、利益相关方的教育、增强内外部沟通、增进监管促进路径的合理使用以及推进监管促进路径全球化协同等。
3.3 完善上市后研究和撤市流程
通过监管促进路径的使用,申办方能够快速将创新药品推向市场。根据科睿唯安新药报告的统计[4],获益最大的2 个治疗领域是抗肿瘤和罕见病。同时,这些加速获批的药物,尤其是通过附条件批准的药物,需要向监管机构承诺上市后研究和持续监测。其中的风险在于,监管机构要求获得完全批准所需的上市后研究结果,与加速审评时提供的小样本研究结果并非总是一致,原因是这些小样本研究往往采用的是替代性终点。因此对于加速批准上市的新药而言,监管机构和申办方均需要重视其上市后研究,以确保可以为患者带来疗效确证、不良反应可控的新药。
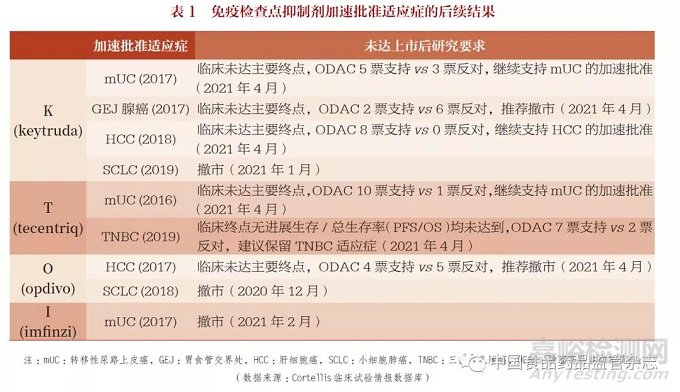
以免疫检查点抑制剂的抗体药物为例,表1 列出了4 种在欧美市场获批的免疫检查点抑制剂,包括keytruda( K药)、opdivo(O药)、tecentriq(T药)和imfinzi (I药)。根据Cortellis 临床试验情报数据库(Cortellis Clinical Trials IntelligenceTM),这4 种免疫检查点抑制剂在2016~2019 年通过加速批准的适应症,在随后的上市后研究中没有达到相应的临床终点,部分直接撤市。另外,部分在2021 年4 月底FDA 的肿瘤药物咨询委员会(ODAC)投票后给出了相应的意见。这也体现了监管机构和申办方对患者负责的严谨态度。
从申办方角度来看,新药产品获批上市能够为企业带来现金流,证明企业开发能力并支持后续的产品管线开发。由于上市后研究可能带来不确定性,作为新药上市的企业,并没有足够的动力开展上市后的验证性临床研究。这里举一个典型案例,来自于己酸羟孕酮(商品名Makena)。该药于2011 年获得批准,是FDA批准的首个用于降低早产风险的药物。批准是基于19 家医院463名早产史女性的使用数据,证实其可降低复发性早产。FDA 批准该药上市的同时,要求申办方开展上市后的验证性临床试验。随后的验证性临床试验纳入了多个国家共计1708 名受试者,在经历了10 年的试验后,其结果未达主要终点,宣告验证性临床试验失败。FDA 在2020 年建议其撤市。Cortellis 竞争情报数据库显示,该企业在这10 年中已经获得超过13 亿美元的销售收入。
由此可以看到,附条件批准或加速批准不是药物研发的终点,药品监管机构需要制定更加完善的后续验证性临床试验的要求,而药企应该积极主动地完成试验承诺,加强药物的全面评估,从而为患者带来真正临床获益的产品。
04药品审评审批的未来关注方向
监管促进路径的设置有效地加速了创新药物获批。但从市场准入到患者可及的过程中,还存在很多亟待解决的问题。时至今日,医药产业的发展,明确了以患者为中心的宗旨。在整个药品研发生命周期中,需要全面考虑和平衡多个利益相关方的诉求,如新药开发商、监管机构、医保支付方、商业保险、患者等,从而真正提高并优化患者可负担的可及性[7]。药品上市不同利益相关方需要回答的核心问题见图8。
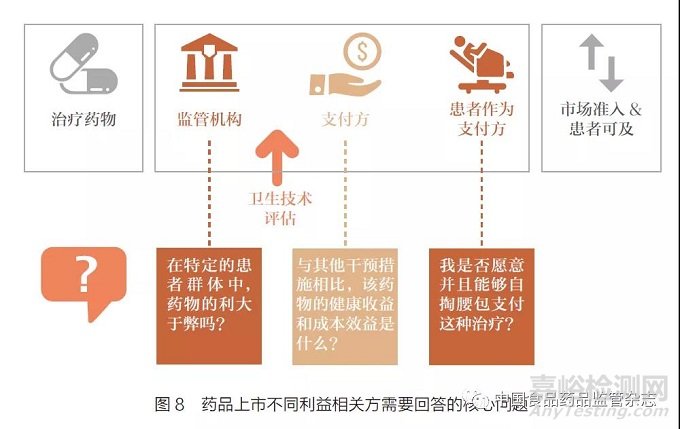
从2015~2019 年欧洲多国、澳大利亚和加拿大的医保准入时间线来看,这几个国家的监管审评所用的时间相差不多,但是从获取上市许可到在卫生技术评估结果的时程上则差异较大。例如,澳大利亚因法规监管和卫生技术评估可以同时进行,从上市许可到卫生技术评估平均仅需几十天,而波兰则平均需要超过500 天[8]。在这些以全民医保为基础的国家中,能否快速获得卫生技术评估的推荐,从而进入医保报销,将决定了该国的患者是否能以较快的速度使用创新药品。
这里以先进疗法为例,包括细胞治疗、基因治疗等。根据科睿唯安CIRS 统计数据来看[9],在EMA 批准先进疗法后,欧洲各国给予的卫生技术评估意见却大相径庭,有推荐、限制性推荐和否定等。其主要原因在于,监管部门与卫生技术评估机构的出发点以及衡量方法各不相同。监管机构是基于严格的临床试验得出的患者获益结果,重点关注新药的收益风险比;而卫生技术评估机构则关注在临床实践中创新治疗方法的实际健康收益,同时还要考量产品相较于本国现有治疗方案的成本效益。
这些具有潜在重大临床价值的药品,有机会获得加速审批或者附条件批准的支持,使目标患者尽早受益。正如前面在申办方对于监管促进路径看法的讨论中提到的,这类创新产品会对卫生技术评估和支付方造成负面影响或矛盾,因为相应的临床证据不够成熟,导致产品在临床疗效和费用方面存在较大不确定性,从而给卫生技术评估机构和医保支付方的评估带来了相应的决策风险。
因此,在获得加速审评的产品(如PRIME 和BTD)开发过程中,其关键临床试验的设计申办方应尽早寻求与监管机构、卫生技术评估机构等利益相关方沟通的机会,以获取支持,提前识别监管审评和卫生技术评估之间证据要求的分歧,从而在后续上市许可申请到医保准入过程中做好准备。
05结 语
全球主要监管机构为应对日趋增长的药品审评审批压力,不断进行大胆尝试,探索出适应各自国家和区域的新方法和新路径。通过关注和比较国外主要监管机构的监管审评发展和未来趋势,可以为我国在新制度下的监管科学研究提供参考。
引用本文
王刚,曾亚莉,雷灿,黄庭颖,Magda Bujar,Ting Wang.监管科学发展下药品审批提效的实践探讨[J].中国食品药品监管.2021.07(210):34-41.
第一作者简介
王刚,博士,科睿唯安生命科学与制药事业部大中华区首席科学家。专业方向:药物研发领域的创新前沿识别、竞争分析

来源:中国食品药品监管杂志


