您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-08-09 21:52
摘要:在仿制药研究过程中通过体外评价方法替代体内生物等效性研究可以减少临床资源消耗,加快药品上市进度,这是基于生物药剂学分类系统(BCS)的生物等效性豁免的主要原因。随着我国仿制药的快速发展,企业基于BCS的生物豁免申请需求越来越迫切,当药物制剂为具有全身作用的普通口服制剂,且药物活性成分符合溶解性和渗透性(BCS I和III类)标准,受试制剂剂型和规格与参比制剂相同,可适用基于BCS的生物等效性豁免。对国内外的基于BCS分类的生物豁免研究进展和法规进行综述,并对具体操作及申报要求提出一些思考。
仿制药研发过程中,口服固体制剂通常需开展生物等效性(bioequivalence)研究,以证明仿制药与原研药具有生物等效性,从而保证其临床的有效性和安全性。生物等效性评价是仿制药质量与疗效一致性评价的重要内容之一。如果研究证明口服固体常释制剂溶解度很高,且在体内的溶出快,药物的吸收速率和吸收程度可不依赖于药物的溶出时间或在胃肠道的通过时间。1995年Amidon等[1]学者提出生物药剂学分类(biopharmaceutics classification system,BCS)的分类概念,建立了药品生物等效性的体外评价方法。依据药物活性成分(active pharmaceutical ingredient,API)的水溶性和肠道渗透性,将其分为4个类别,即Ⅰ类(高溶解性、高渗透性)、Ⅱ类(低溶解性、高渗透性)、Ⅲ类(高溶解性、低渗透性)和Ⅳ类(低溶解性、低渗透性)。对于BCS I类和III类的药物,只要处方中的其他辅料成分不显著影响API的吸收,且非窄治疗窗或口腔吸收等药物,则不必证明该药物在体内生物利用度和生物等效的可能性,即生物等效性豁免(waiver of in vivobioavailability andbioequivalence)。制剂之间的治疗等效性一般需要进行体内生物等效性研究来论证,但是如果体外研究能够充分证明体内性能无差异,那么可以豁免此项研究。仿制药更改剂型和剂量时,如何减少耗时耗资源的体内研究,仅采用测定体外溶出度方法来进行药品上市后的变更以及放大的放行,这就是基于BCS豁免生物效性研究的最初考虑[2]。当剂型符合设定的标准时,申请人和监管机构可依据BCS评判该制剂是否可以豁免生物等效性研究,这就是基于BCS的生物等效性豁免的定义[3]。
基于BCS分类的生物等效性豁免方法为部分仿制药提供了豁免生物等效性研究的途径。根据BCS的基本理论,影响普通口服固体制剂API吸收速度和吸收程度的主要因素是API的BCS类别与药物制剂的溶出特征和辅料组成。
生物等效性豁免经过不断地发展,主要适用于口服给药的速释固体剂型或混悬剂,并且豁免的药物大多属于BCS Ⅰ和Ⅲ类[2]。随着我国仿制药的发展,企业基于BCS分类的生物豁免申请需求越来越迫切,而目前我国基于BCS分类的生物豁免申请还处于起步阶段,虽然已有生物豁免申报案例,但申办方往往对该项研究比较陌生。为了更好推动该项研究,本文对国内外的基于BCS分类的生物豁免研究进展和法规进行比较,并对具体操作及申报要求提出一些思考。
1、各国关于基于BCS分类的生物等效性豁免的新进展
1.1FDA基于BCS分类系统的生物等效性豁免新进展
20世纪90年代,美国食品药品监督管理局(FDA)以及学术界一直致力于建立1种避免不必要人体生物等效性研究的科学方法。2000年8月FDA发布了免除体内生物等效性试验的行业指导原则[4],2000年版BCS指导原则指出,若BCS Ⅰ类药物的口服固体常释制剂具有快速体外溶出,且与参比制剂的溶出曲线相似,则可以豁免其人体生物利用度或生物等效性研究[5-7]。
2003年制定的BCS为仿制药品的开发提供了一条新途径[2]。此后多年,FDA不断对3个关键问题进行研究和讨论,即①高渗透性的标准可否降低;②溶解性试验中pH值的范围可否缩窄;③生物豁免可否扩展至BCS Ⅲ类药物[5,7-8]。
FDA于2017年12月22日发布了根据BCS豁免速释固体口服制剂体内生物利用度和生物等效性研究指导原则正式版本[6]。与2000年版相比,2017年版BCS指导原则包含了3项重要修订,即①高渗透性的标准由90%降至85%;②溶解性试验中pH值由1.0~7.5缩窄至1.0~6.8;③生物豁免扩展至BCS Ⅲ类药物,但对其制剂的溶出和辅料等提出更严格的标准[9]。
1.2 世界卫生组织(WHO)基于BCS的生物等效性豁免最新进展
在FDA相关工作的基础上,WHO于2006年发布《仿制药:建立可替代性的注册要求指导原则》,普通口服固体制剂仿制药(BCS Ⅰ、Ⅲ类和部分Ⅱ类API)在特定条件下可豁免生物等效性研究,同时发布2006年版豁免名单[10]。2015年WHO发布了新版《仿制药:建立可替代性的注册要求指导原则》,并于2017年重新发布补充附录。2017年版指导原则提出了BCS Ⅱ类API不再适用基于BCS的生物等效性豁免[11]。此外WHO在2018和2020年均对药物生物等效性清单进行了更新,重新评估了豁免清单中的药物是否适用基于BCS的生物等效性豁免并提出相关建议,2020年公布了16个API的评估结果,此次评估的结果更加准确可靠[12]。
随着新冠疫情的爆发,如何快速审评具有抗新冠肺炎并发症的药物是药物可及性的一个关键,基于BCS分类的生物豁免研究尤为重要。
1.3 欧洲药品管理局(EMA)基于BCS的生物等效性豁免最新进展
2010年EMA出台了《Guideline on the investigation of bioequivalence (CPMP/QWP/EWP/1401/98Rev.1,2010)》[13]。欧盟对生物等效性豁免研究是基于生物等效性研究指南,同时也和ICH出台的政策相适应。
1.4 国际人用药品注册技术协调会(ICH)基于BCS的生物等效性豁免最新进展
为了加快新药上市并保证已批准药物能够持续对患者可及,避免在人体上重复开展临床试验,ICH通过在国际层面上协调技术要求,致力于推动药品注册技术要求的国际统一,旨在能更经济有效地研发、注册和生产高质量的安全有效药物[14]。基于该目的,ICH大会于2016年10月通过“M9:Biopharmaceutics Classification System-Based Biowaiver”议题。2019年11月ICH正式发布指导原则《M9:基于生物药剂学分类系统的生物等效性豁免》。自M9进入实施阶段后,欧洲一些主要的管理机构于2020年7月30日正式实施,接着加拿大和瑞士于同年8月正式实施,中国、美国及日本均处于实施过程中[15]。
1.5 我国基于BCS的生物等效性豁免最新进展
2016年5月国家原食品药品监督管理总局发布《人体生物等效性试验豁免指导原则》。此外可豁免或简化人体生物等效性试验品种第一批清单和第二批征求意见稿也分别于2018年5、7月发布,同年7月发布的征求289种基本药物目录中的国内特有品种评价建议,有62个品种有望通过生物等效性豁免申请简化评价流程,可以大大减少不必要的健康人体试验,从而加快评价速度和降低评价费用[16]。国家药品监督管理局(NMPA)于2018年12月发布了“可豁免或简化人体生物等效性试验品种”的通告,其中包括5个品种可根据上述豁免指导原则申请基于BCS的生物等效性豁免[17]。
2、FDA、WHO、EMA、ICH、NMPA关于生物等效性豁免的差异性
FDA是最早提出生物等效性豁免指导原则的机构,并且不断根据发展更新了指导原则,其对于豁免的范围主要为BCS I和Ⅲ类,对BCS Ⅲ类的药物有更多限制条件。对于快速溶出的BCS Ⅲ类药物而言,WHO特别指出应从药物的吸收部位、吸收机制和处方中辅料组成以及治疗风险角度进行更谨慎的风险评估。
另外,对于不同BCS分类药物的体外溶出测试,WHO也提出了明确要求。对于BCS I类的药物而言,体外溶出标准同FDA,但调整了桨法的常规转速(由50 r/min调整到75 r/min)。考虑到渗透性是药物体内吸收的限速步骤,对于BCS Ⅲ类的药物而言,药物在生理条件下能够快速溶出(类似口服溶液剂),故要求药物在pH值1.2、4.5、6.8缓冲盐体系中,在常规的溶出度测定方法(桨法75 r/min或转篮法100 r/min)下于15 min内的溶出超过85%。
对于渗透性的要求,EMA给出了比FDA更为具体的要求。EMA强调渗透性数据应来自人体研究,规定应根据可靠的人体研究和合理质量平衡研究来得到数据。在使用质量平衡研究数据来支持药物的完全吸收特性时,如果将代谢物也纳入药物吸收量的计算,研究者应确保该代谢物是在胃肠道吸收后才形成的。
我国内发布的《人体生物等效性豁免指导原则》主要参考FDA指导原则制定,同时参考了《中国药典》2015年版、《美国药典》39版以及WHO和EMA的相关技术要求。下面主要从溶解度、渗透性、溶出度测定等几个方面来比较,见表1、2、3。
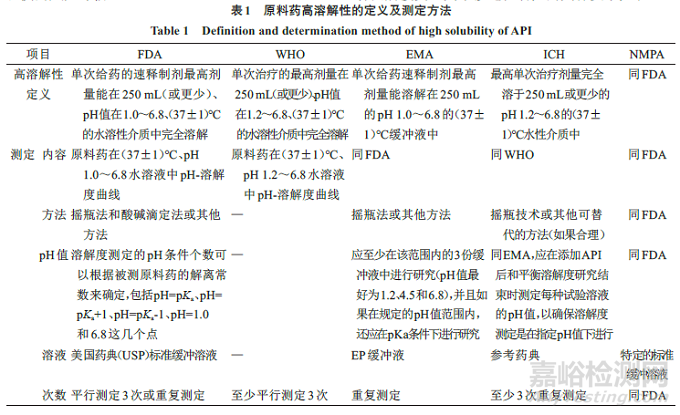

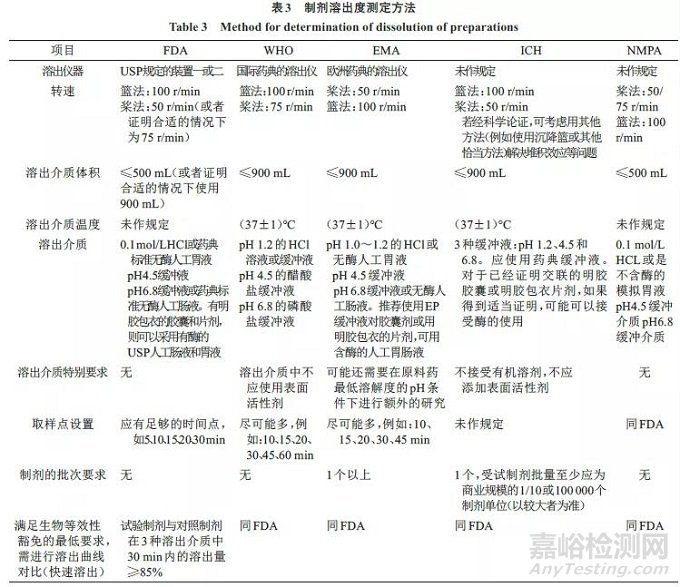
ICH M9指导原则是ICH专家在各国相关指导原则技术要求基础上综合而来,旨在减免不必要的浪费,加快药物的研发速度,降低研究成本,因此其政策逐渐被各国所接受。其中渗透性研究的具体要求见表4。

从溶解度研究的角度而言,虽然FDA规定的pH值在1.0~6.8和WHO规定的pH值在1.2~6.8有一定差异,但具体操作差异性不大。如FDA规定的包括pH=pKa、pH=pKa+1、pH=pKa-1、pH=1.0、6.8,WHO未作具体规定,而EMA和ICH规定pH值在1.2~6.8,至少测定3个pH值:1.2、4.5和6.8,如果pKa落在该范围内,需要另外增加该pH值的溶解度。我国NMPA遵循了FDA的指导原则,同时参考ICH指导原则,申办方在具体操作中也应根据药物特性选择合适的pH值进行实验,以更好地体现药物溶解特性。虽然文献数据也可作为生物豁免申请的支持材料,但考虑到论文可能无法保证数据的质量,为了更好地评价符合生物豁免的品种,建议NMPA设定专门的实验室进行该项研究,可以减少研发成本,提高研究效率。
不同实验室从事体外和体内渗透性研究时,模型药物的结果存在室间差异,为了提高结果的可靠性,不同实验室应建立自己的模型药物数据库和判断指标,例如高渗透性的判定值等。此外,在Caco-2细胞研究中,对不同批次的细胞应进行考察,以确保实验结果不会受到不同批次细胞差异导致的实验假阳性。在今后的政策制定过程中,建议相关部门综合参考FDA和ICH的指导原则进行修改。
3、结语
通过研究者和政策制定者的不断努力,基于BCS的生物豁免研究将被更多申办方和审评人员接受。今后在生物豁免的研究中,不断细化研究细则,不断加强人员培训。体外溶出方法的开发还应结合体内药动学和体内外相关性分析进一步优化,可以为今后我国生物豁免的研究和审评提供更多的理论支持,加快我国仿制药的研究速度。
参考文献(略)

来源:Internet


