您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-06-24 22:52
相容性研究背景
药品包装应满足药品预期目的的适用性要求。适用性包括了保护性、功能性、安全性和相容性。《药品管理法》明确指出“直接接触药品的包装材料和容器,应当符合药用要求,符合保障人体健康、安全的标准”。
药品与其直接接触的包装系统满足相容性要求,是保证药品质量的安全必须具备的条件之一。相容性研究则是评价包装组件或系统与药品直接接触后,没有发生严重的、或不可接受的导致药品有效性和稳定性发生改变,或者产生安全性风险的过程,并;研究内容包括包装材料或组件的可提取研究、药品与包装系统接触的影响的浸出研究和可能产生的对药品和辅料活性成分的吸附研究。
药品包装对于保证药物稳定性起着重要的作用,因而将直接影响药品使用的安全性。直接接触药品的包装材料、容器是药品的一部分,由于包装材料、容器组成配方、所选择的原料及生产工艺的不同,导致不恰当的材料中添加剂和配方成分的迁移,或吸附有效成分、甚至发生化学反应,使药物失效,有的还会产生严重的副反应。这就要求在为药品包装材料之前,必须检验正式是否适用于预期用途,必须充分评价其对药物稳定性的影响,评价其在长期的储存过程中,在不同的环境条件下(如温度、湿度、光线等),在运输过程中(如与药物接触反应、对药物的吸附等)、容器(材料)对药物保护效果和本身物理、化学、生物惰性,所以在选择和使用药包材之时必须要做相容性研究。
法规要求
1,ChP 2015,四部9621,药包材通用要求指导原则;
2,ChP 2015,四部9622,药用玻璃材料和容器指导原则;
3,化学药品注射剂与塑料包装材料相容性研究技术指导原则(试行)(国食药监注〔2012〕267号);
4,化学药品注射剂与药用玻璃包装容器相容性研究技术指导原则(试行)(国食药监通告2015第40号);
5,化学药品与弹性体密封件相容性研究技术指导原则(试行);
6,原料药、药用辅料及药包材与药品制剂共同审评审批管理规定;
7,ICH M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk;
8,ICH Q3D Guideline for Elemental Impurities;
9,EMEA Guideline on Plastic Immediate Packaging Materials. (2005)
10,European Pharmacopoeia (Ph.Eur)Chapter 3 “Materials and Containers”.
11,USP <660><661><671><1660><1661><1663><1664>;
包材的风险程度
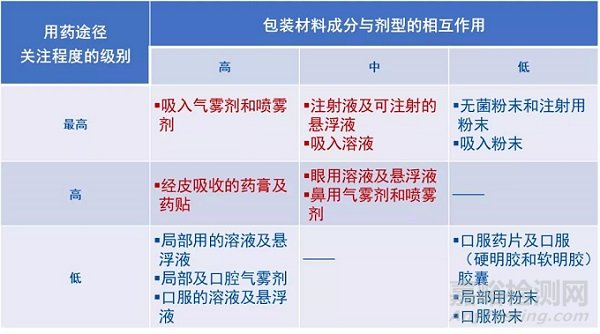
是否需要进行相容性研究,以及进行何种相容性研究,应基于制剂的给药途径的风险类型与直接接触的包装发生相互作用的可能性进行评估,进行确认。美国FDA指南认为吸入制剂和注射用药等的风险较大,需要进行的研究和提供的资料最为详细。
风险相对较小的固体口服制剂等剂型中,大部分塑料容器的数据可以参考食品添加剂的(21 CFR 174-186)要求。
研究的总体思路是药物与包装材料发生相互作用的可能性和药物的给药途径对人体安全的风险。
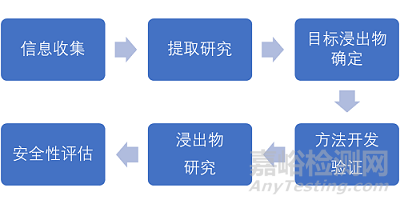
相容性研究流程
(CDE指南上有六步法,请参考)
包材信息收集
在做药包材相容性检测时,首先需要确定直接接触药品的包装组件及成分。比如塑料类药包材成分有聚乙烯、聚丙烯、环状聚烯烃、聚碳酸酯等种类,而玻璃类药包材成分有二氧化硅、三氧化二硼、三氧化二铝、氧化钠、氧化钾、氧化钙、氧化镁等种类。不同材料的药包材成分自然不一样的,确定好药包材的具体配方成分,这也是药包材检测需要了解的基本概念。
包材相容性研究,需要了解或分析包装组件材料的组成部分,药包材的组件很多情况不是单一的,比如一些注射剂药瓶,密封件是金属铝制的,瓶塞是橡胶的,瓶身是玻璃的。所以一定要对各个组件进行详细了解,除此之外,包装组件与药品的接触方式与接触条件以及药包材的整个生产工艺过程,都需要面面俱到的考察清楚,然后把相关资料提供给检测方。

欧洲药典收载了聚丙烯、聚乙烯材料通常使用的添加剂,包括结构、名称、最低限量和测定方法,企业在进行药物相容性试验时可参考。
提取物和浸取物研究
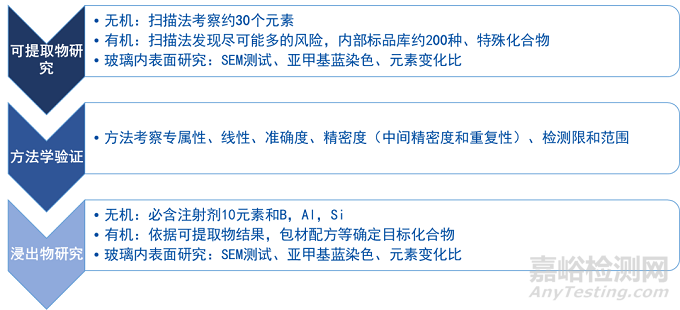
提取研究对于药包材相容性研究极其重要,提取研究主要有药包材的样品处理、提取溶剂的选择、提取条件的确定等。通过提取研究,尽可能获得更多的可提取物,对获得的相关可提取物进行鉴别、定量,预测药包材潜在的可浸出物,这其中包括单体、起始物质、残留量、降解物质、分子量低于1000D的物质的添加剂或助剂等的检查。
完成上述检查后,进入到药包材相容性检测的核心部分,对试验药品制剂与包装材料的相互作用研究。相互作用研究主要是通过迁移试验和吸附试验来实现的。在做迁移试验时,应该充分考虑药品在生产、贮存、运输及使用过程中可能面临的最极端条件,尽量选择药品上市后最高浓度,并且在加速及长期稳定性试验条件下进行。而吸附试验应该注意温度和时间的选择上,因为吸附试验对活性成分和辅料的检测,所以可以适当的增加检测指标,例如辅料含量、pH值等。
通过提取研究及相互作用研究,就可以得到药包材和可提取物、浸出物及吸附效果的信息,通过这些信息,便可以分析药包材与药品是否会产生浸出物,是否对药效产生影响,是否会对药物或辅料产生吸附效果,从而影响药品质量等等问题。将这些问题汇总分析,来对药包材的安全性进行风险评价。从而得出药包材是否与药品具有相容性。
常用分析技术
模拟试验和迁移试验应采用专属性强、准确、精密、灵敏的分析方法,以保证试验结果的可靠性;并应针对不同的待测项目选择适宜的分析方法。由于玻璃容器最常见的可提取物为金属离子、不挥发性物质等组分,对可提取物和浸出物的常见分析方法包括:电感耦合等离子体发射光谱(ICP)、原子吸收光谱(AAS)、离子色谱(IC)、高效液相色谱(HPLC),以及与质谱的联机技术,ICP-MS、HPLC-MS等,方法学研究时重点关注灵敏度(检测限、基线值)、专属性、准确性等。

安全性评估
依据FDA、CFDA、ICH、欧洲药品局(EMA)、致癌性数据库(CPDB)、美国毒物和疾病管理中心(ATSDR)数据库、美国环境保护署(EPA)数据库等标准对迁移物/浸出物进行安全性评估。
根据浸出物的PDE值、每日最大用药剂量计算每单个包装容器中各浸出物的最大允许浓度,并在此基础上经计算得到分析评价阈值AET,分析测试方法应满足该AET值的测定要求。如果迁移试验显示浸出物含量低于PDE时,可认为浸出物的量不会改变药品的安全性,对患者的安全性风险小。如果迁移试验显示浸出物的含量高于PDE,则认为包装容器与药品不具有相容性,建议更换包装材料。
对于玻璃包装,除了关注浸出物的风险,还要关注药品对玻璃内表面的影响,确认没有脱片的风险。
如果试验发现出现溶液颜色加深、产生可见异物、pH值变化等现象,应分析原因并对试验结果进行评估。如果上述变化已达到不可接受的程度,且为玻璃容器所致,应考虑采用其他类型玻璃包装容器以及其他形式的包装容器;如果是其他原因所致,应对产品进行优化,如完善制剂的处方工艺等以使产品符合相关质量控制要求。
如果吸附试验结果显示包装容器对药品或辅料存在较强吸附,并对药品质量产生了显著影响,建议采用适宜的方法消除对产品质量的影响,比如,更换包装容器。
影响药包材相容性检测的重点因素有药包材类型、组成部分、规格大小、药包材处理方式、药品的性质(PH值、离子强度)、生产工艺等,药包材在药品生产过程中的清洗、灭菌的处理,如玻璃容器在洗瓶阶段的干热灭菌、制剂冷冻干燥、终端灭菌都需要纳入药包材相容性检测的考察范围之内。
名词注释
分析评价阈值(Analytical Evaluation Threshold, AET):其含义是在这个阈值以上的可提取物和(或)浸出物均应定性及定量,并报告同时进行潜在的毒性评估。
根据人每日允许最大暴露量或安全性阈值/限定阈值、用药剂量以及制剂包装特点等计算每单个包装容器中特定的可提取物和/或浸出物含量,当一个特定的可提取物和/或浸出物水平达到或超过这个量值时,需要开始对这个可提取物/浸出物进行分析,并需要报告给相关部门以便开始进行安全性评估。
安全性阈值(Safety Concern Threshold, SCT):其含义是只要低于这个阈值,浸出物无论是否具有致癌性,均可忽略其安全性风险。SCT是绝对暴露量,每日的总摄入量;进行相容性研究时将其转化为特定药品中单个浸出物的浓度限值。
AET估值:可由SCT估算;先将SCT(μg/日)转换为产品单位剂量中的量,例如,μg/罐、μg/剂、μg/粒;再根据产品中各组件的质量和数量将这个值转换为每克组件中的量,例如,μg/g,即产品中的量与组件质量的比值;此结果就是AET估值。分析方法的灵敏度(定量限LOQ)要求AET估值可被定量测定。
AET终值:应对用于确认可提取物与浸出物谱的分析方法的不确定性进行估计。估计不确定性的一种方法是建立一个通过可靠的标准品(可获得的)得到的可提取物响应因子(RF)的数据库。对方法的不确定性进行的评估用于由AET估值计算出AET终值。该计算过程允许分析化学家修订原始的AET估值;如有必要,也可对未经评估的可提取物与浸出物进行鉴定。
人每日允许最大暴露量(permitted daily exposure, PDE):指某一物质被允许摄入而不产生毒性的日平均最大剂量,某一具体物质的PDE值是由不产生反应量、体重调整系数、种属之间差异的系数、个体差异、短期接触急性毒性研究的可变系数等推算出的。
界定阈值(Qualification Threshold,QT):其含义是在这个阈值下如果浸出物是非致癌性物质,则无需进行安全性界定(毒理学评估),除非浸出物存在结构活性风险。
相容性研究:包装系统与药物相容性研究是指考察包装系统与药物之间是否发生迁移或吸附等, 进而影响药物质量和安全性而进行的试验过程。
可提取物:通过提取试验获得的从包装材料中溶出的物质。
浸出物:通过迁移试验获得的从包装系统中迁移或因此而产生的并进入至药品中的物质。
玻璃内表面耐水性:是一种表面试验法,是用规定的水注入被测容器到规定的容量,并在规定温度、时间下进行处理后,通过滴定浸提液来测量水对容器内表面的侵蚀程度。
玻璃内表面耐受性:指玻璃容器在其包装内容物期间,内表面承受水、酸、碱等物质的物理、化学侵蚀以及温度、压力等环境因素作用的力。
脱片:玻璃内表面的碱金属离子受溶液化学侵蚀,在玻璃表面形成一层高硅氧层。高硅氧层与玻璃内部的未变质玻璃膨胀系数不同,在温度变化时两者之间会产生应力,导致高硅氧层从主体玻璃上脱落到溶液中形成玻璃脱片的现象。除上述原因以外,也可能存在其他导致玻璃脱片的因素。

来源:Internet


