您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-03-16 09:41
摘要
蛋白降解靶向嵌合体(PROTAC)技术是一种通过泛素 - 蛋白酶体途径化学诱导靶蛋白降解的新兴策略。PROTAC 是由靶蛋白配体和 E3 泛素连接酶配体通过适当的连接链连接而成的双功能分子,能够同时招募靶蛋白和 E3 泛素连接酶,从而诱导靶蛋白泛素化降解,具有广泛的应用前景和发展空间。主要简述小分子 PROTAC 的降解机制、发展历程,以及在药物研发中面临的机遇与挑战。
自然界已进化出高效的酶系统来调控泛素化和随后的蛋白质降解。如果这些酶系统能够被人为利用和重新靶向以达到疾病的治疗目的,其潜力将是巨大的。蛋白降解靶向嵌合体(proteolytic targeting chimera,PROTAC)技术正是一种人为化学诱导靶蛋白(protein of interest,POI)多聚泛素化,最终通过蛋白酶体通路靶向降解 POI 的新兴技术,为疾病的治疗提供了新的策略。PROTAC 是由靶蛋白配体和 E3 泛素连接酶配体通过适当的连接链组成的双功能分子,能够同时招募靶蛋白和 E3 泛素连接酶诱导靶蛋白泛素化降解,其独特的作用模式具有广泛的应用前景和发展空间,备受学术界和制药工业界的关注,在克服耐药性以及传统“不可成药”(undruggable)靶点方面有巨大的潜力,更是一种全新的药物研发策略 [1]。
01、PROTAC 的降解机制及特点
通常,细胞内蛋白质通过泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)和自噬 / 溶酶体 2 条途径进行降解 [2]。2004 年诺贝尔化学奖被授予 Ciechanover、Hershko 和 Rose 这 3 位科学家,以表彰他们在泛素调节蛋白质降解方面的卓越贡献, 此后泛素介导的蛋白降解机制被稳步揭晓。蛋白质泛素化是一种多功能的蛋白质翻译后修饰过程,影响着细胞的分化、增殖、转移、凋亡、基因表达、信号传递等整个生命过程。泛素化过程可以简单地概括为:泛素标签首先与 E1 泛素激活酶结合,然后转移到 E2 泛素结合酶上,随后依赖于一个大家族的衔接蛋白(E3 泛素连接酶)将其泛素传递到靶蛋白上。被泛素标记的蛋白质被蛋白酶体特异性识别并降解 [3]。蛋白酶体是细胞内的一种复合物,主要负责将特异性泛素化的蛋白质降解 [4](见图 1)。
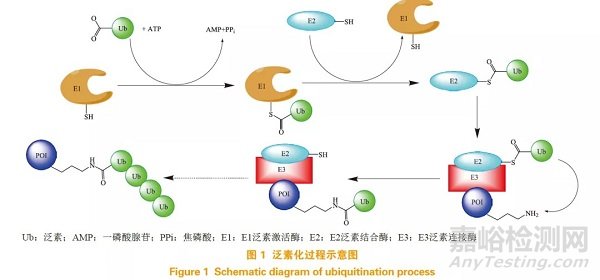
PROTAC 分子由靶蛋白配体、连接链和 E3 泛素连接酶配体 3 部分组成,是一种能特异性结合靶蛋白同时招募 E3 泛素连接酶并使 POI 多聚泛素化、最终通过蛋白酶体系统降解的双功能分子 [5]。
PROTAC 分子泛素化标记 POI 并促使蛋白降解后可与 POI 解离并可在细胞内循环利用,起到亚化学计量催化的效果,减少药物的使用剂量从而减少体内药物暴露量,降低毒副作用 [6](见图 2)。
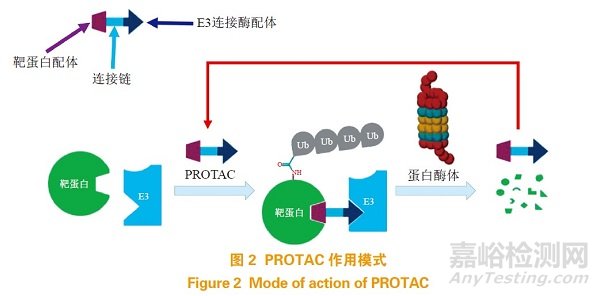
从作用模式来看,PROTAC 分子与传统的抑制剂有着根本的区别。传统的抑制剂是以占用驱动(occupancy-driven)的作用模式,特异性结合于靶蛋白的空腔内。这种模式需要较高的药物浓度, 以维持对靶蛋白的占用水平,进而发挥药理活性, 获得临床应用价值 [7]。相反,PROTAC 是事件驱动(event-driven)的作用模式,其不受均衡占有率(equilibrium occupancy)的影响,在较低浓度就能够实现超 90% 的靶蛋白降解,这对占用驱动模式是难以实现的 [8]。
02、PROTAC 重要发展历程
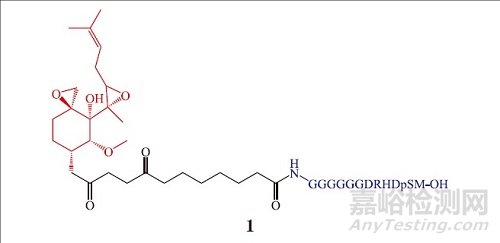
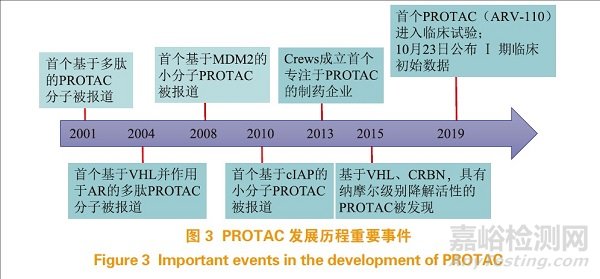
早在 2001 年, 耶鲁大学 Craig M. Crews 教授团队和加州理工大学的 Raymond J. Deshaies 教授首次提出 PROTAC 这一概念并经过一系列的概念验证(proof of concept)报道了首个 PROTAC 分子——Protac-1(1),其可靶向降解甲硫氨酰氨肽酶-2(MetAp-2)[9]。随后继续对其深入研究, 不断扩大应用范围并成功降解雌激素受体(ER)和雄激素受体(AR)[10]。
基于希佩尔·林道(Von Hippel Lindau,VHL)E3 泛素连接酶的多肽类 PROTAC 于 2004 年被首次报道 [11]。这些早期基于多肽类的 PROTAC 是源于转录因子低氧诱导因子-1α(HIF-1α)衍生的肽序列构建的,HIF-1α 被脯氨酰羟化酶羟化后能与 VHL 蛋白结合。随后的大量研究工作发现了阻断 HIF-1α 和VHL 相互作用的小分子抑制剂 [12]。X 射线衍射技术阐明了高效小分子抑制剂的结合模式。最初的 HIF-1α 衍生肽被含有羟脯氨酸结构的小分子取代,得到了高亲和力和高特异性 VHL 配体[12]。大量文献研究表明,基于小分子的 VHL-PROTAC 能有效降解雌激素相关受体(ERRα)[6]、AR[13]、激酶 RIPK2 [6]、BCR-ABL 融合蛋白 [14-15]、含溴区结构域蛋白质(BRD)4 [16]、TBK1、跨膜酪氨酸激酶(EGFR、HER2 和 c-Met)[17]、MEK[18] 和 TRIM24 蛋白 [19]。
早期基于多肽的 PROTAC 在体外验证以及作为生物化学分子工具具有一定的价值,然而由于细胞通透性、化学稳定性以及成药性方面的问题,其在体内的进一步研究受到一定程度限制。因此,非肽类 PROTAC 在该领域的研发至关重要。
2008 年,耶鲁大学 Craig M. Crews 教授团队首次合成了含有 nutlins 的小分子 PROTAC,成功地将 AR 募集到鼠双微体 2(mouse double minute 2,MDM2)上,并作为 E3 泛素连接酶触发其泛素化和蛋白酶体降解 [20]。MDM2 是一种主要靶向肿瘤抑制因子 p53 的 E3 泛素连接酶 [21]。此外,Craig M.Crews 团队的研究结果表明,以 idasanutlin 为 MDM2配体,JQ1 为 BRD4/BET 抑制剂组成的 MDM2 诱导BRD4 泛素降解的 A1874 化合物对靶蛋白 BRD4 的降解率为98%,在纳摩尔水平表现出较好的生物活性[22]。值得注意的是,该 PROTAC 既能降解 BRD4 又能稳定 p53,是关于 E3 泛素连接酶配体和靶蛋白配体的协同抗增殖作用的首次报道。2019 年 1 月,清华大学饶燏教授团队基于 MDM2 的 E3 泛素连接酶设计并合成了具有靶向降解作用的多腺苷二磷酸核糖聚合酶(PARP1)降解子 [23]。
2010 年,Ito 等 [24] 揭示了沙利度胺致畸作用的分子靶点和主要原因——Cereblon 蛋白(CRBN)。沙利度胺及其衍生物来那度胺和泊马度胺作为免疫调节药物(IMiD)[25] 已被批准用于多发性骨髓瘤。在机制上,IMiD 靶向 E3 泛素连接酶 CUL4- RBX1-DDB1-CRBN(也被称为 CUL4CRBN)。IMiD 与 CRBN 结合,使得 IKAROS 家族转录因子(IKZF1 和 IKZF3)被募集到 CRBN 上进而泛素化降解 [26]。2014 年,与沙利度胺和来那度胺结合的DDB1-CRBN 复合物晶体结构得以解析 [27],从此, 以 CRBN 为靶点的 IMiD 小分子和以各种蛋白质为靶点的 PROTAC 迅猛发展。现已成功开发出靶向BRD2/3/4 [28]、FK506 结合蛋白 12(FKBP12)[29]、BCR-ABL 融合蛋白[30]、BRD9 [31]、沉默信息调节因子2(Sirt2)[32]、细胞周期蛋白依赖性激酶 9(CDK9)[33]、FMS 样酪氨酸激酶 3(FLT3)[34]、布鲁顿酪氨酸激酶(BTK)[35]、间变性淋巴瘤激酶(ALK)[36]、周期蛋白依赖性激酶 4/6(CDK4/6)[37] 和组蛋白去乙酰化酶 6(HDAC6)[38] 的小分子 PROTAC。
另一种 E3 泛素连接酶,细胞凋亡抑制蛋白 1(cIAP1)也被用于 PROTAC 的设计。2010 年,Hashimoto 课题组公开了由甲基贝他定(methyl bestatin,MeBS)和全反式视黄酸(ATRA)组成的 PROTAC,其中 MeBS 可选择性地结合到 cIAP1 的 BIR3 结构域(其 RING 结构域促进自动泛素化),维甲酸受体内源性配体 ATRA 能募集细胞内维甲酸结合蛋白 CRABP-1 和 CRABP-2[39]。该项工作是首次基于 cIAP1 的 PROTAC 成功地诱导CRABP-1 和CRABP-2 靶蛋白泛素化蛋白酶体降解。通过用 MV1 配体(即 cIAP1/cIAP2/XIAP 配体)替换 MeBS,可进一步改进 PROTAC,实现 cIAP1 和CRABP-2 这 2 种蛋白同时敲除 [40]。其他开发的该类 PROTAC 小分子还有 SNIPER(specific and non- genetic IAP-dependent protein eraser),该小分子可靶向降解 ERα [41]、BRD4[42]、转录相关酸性卷曲蛋白 3(TACC3)[43] 和 BCR-ABL 融合蛋白 [44]。
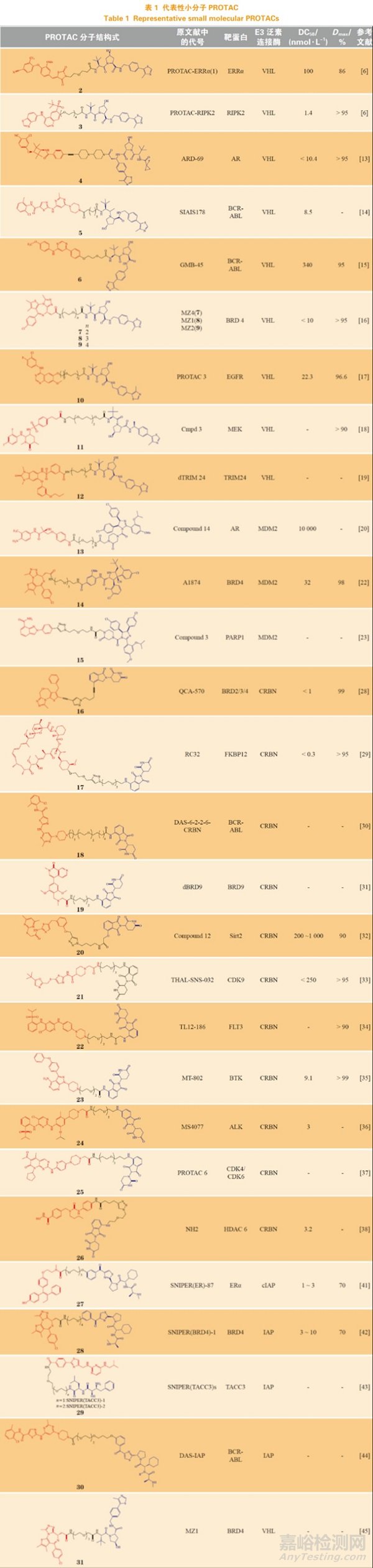
2017 年,Ciulli 教授领衔的科研团队首次报道了 PROTAC 分别与 E3 泛素连接酶和靶蛋白受体形成的稳定三元复合物共晶结构 [45]。实验证明,PROTAC 分子是通过中间连接链适当地折叠弯曲,让其两端分别深入两受体疏水空腔内部,拉近 E3 泛素连接酶与靶蛋白的距离,形成特异性的蛋白 - 蛋白相互作用,促进靶蛋白的泛素化。该研究为后续的理性 PROTAC 药物分子设计奠定了基础。PROTAC 发展历程中的重要事件见图 3。表 1 介绍了具有代表性的小分子 PROTAC(2 ~31)。
03、小分子 PROTAC 新药研发面临的机遇、挑战及研究近况
PROTAC 技术问世至今已经走过近 20 年的发展历史,据不完全统计,现已有超过 30 个靶点可以被PROTAC 分子泛素化降解 [46],展现出广阔的疾病治疗前景。自 2013 年起,Arvinas、C4 Therapeutics、Kymera Therapeutics 等专注于 PROTAC 分子开发的公司相继成立,默克、基因泰克、辉瑞、诺华、勃林格殷格翰等制药巨头也纷纷布局这一技术领域。制药工业界的加入必将给 PROTAC 带来新的发展契机。2019 年 3 月,Arvinas 公司宣布其开发的首个用于治疗前列腺癌的 AR 降解剂 ARV-110 进入Ⅰ期临床试验(NCT03888612)。2019 年 10 月 23 日,Arvinas 公司公布了 ARV-110 和 ARV-471 的Ⅰ期临床试验初始结果:口服 PROTAC 对肿瘤患者具有良好的安全性和耐受性。ARV-110 是公开报道的首个进入Ⅰ期临床试验的 PROTAC 分子,标志着PROTAC 技术的研究进入新的阶段。鉴于 PROTAC 的分子特点及其独特的作用机制,PROTAC 技术在新药研发及疾病治疗中面临着诸多机遇与挑战 [47]。
3.1 PROTAC 的机遇
3.1.1 有望克服耐药性
肿瘤耐药已成为当前临床治疗面临的主要挑战。随着新靶点和新型药物发现技术不断涌现,通过小分子药物靶向致病蛋白或受体为肿瘤的治疗提供了强有力的策略。特别是在过去的 20 年里激酶抑制剂领域蓬勃发展,在临床应用中取得了惊人的效果,极大地改善了肿瘤患者的生活质量,并延长其生存期 [48]。然而尽管靶向药物的效果十分显著,但患者在治疗一段时间后往往会出现不同程度的耐药,造成疾病复发。因此开发新技术克服肿瘤耐药是抗肿瘤研究的热点。
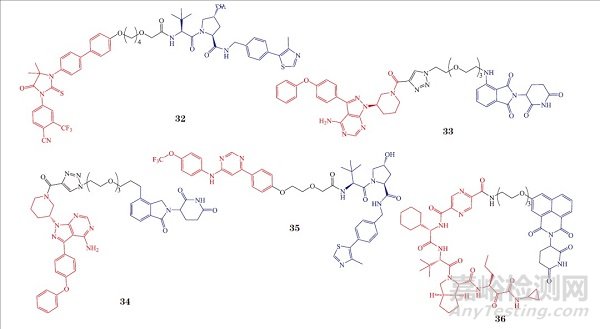
经过近 20 年的发展,PROTAC 在克服肿瘤耐药性方面已获得显著性进展 [49]。2018 年 Craig M. Crews 课题组研发的靶向 AR 的PROTAC 分子 ARCC-4(32) 可有效克服前列腺癌对恩扎鲁胺的耐药性(DC50 = 5 nmol · L-1,Dmax = 98%)[50]。清华大学饶燏课题组开发的 PROTAC 分子 P13I(33)可以同时降解野生型BTK(DC50 = 9.2 nmol · L-1,Dmax = 89%)和对 ibrutinib 耐药的 C481S 突变的 BTK(DC50 = 30 nmol · L-1)[51]。该课题组开发的第 2 代 PROTAC 小分子L18I(34) 水溶性显著提升且可降解不同 C481 突变的 BTK(DC50 < 50 nmol·L-1)[52]。PROTAC 分子 GMB-475(35)不仅可以降解野生型 BCR-ABL 还能降解特定突变的 BCR-ABL[15]。GMB-475 是基于 imatinib 的新颖 PROTAC 分子,在 300 nmol · L-1 下对 K562 和 Ba/F3 细胞的 BCR-ABL1 和 c-ABL1 均有显著降解作用。GMB-475 对BaF3(T315I 突变)的 IC50 达1.98 μmol · L-1,活性是 imatinib 的 20 倍,对 G250E 突变的细胞株 IC50 达 0.37 μmol · L-1。在降解方面, GMB-475 可以完全降解 G250 突变蛋白(DC50 = 310 nmol · L-1), 部分敲低 BCR-ABL1 T315I 蛋白。PROTAC 技术不仅在克服肿瘤耐药性方面获得重大进展,也有望被用于解决其他临床疾病的耐药性问题。Priscilla L.Yang 课题组设计并合成了首个可有效降解病毒蛋白的 PROTAC 分子——DGY-08-097(36)(DC50= 50 nmol · L-1)[53]。该研究工作成功证明可通过降解病毒蛋白的新策略解决病毒抑制剂的耐药问题。
3.1.2 提高靶向选择性
在结构优化中,增加化合物对靶蛋白的选择性是药物研发的重要目标。药物在体内的作用环境是复杂的,有许多潜在的因素会与之发生作用。蛋白质、DNA、RNA、脂类、糖类、代谢物和其他小分子化合物都有可能与药物相互作用 [54]。在许多情况下,这种相互作用会导致意想不到的生化反应,甚至是毒副作用。
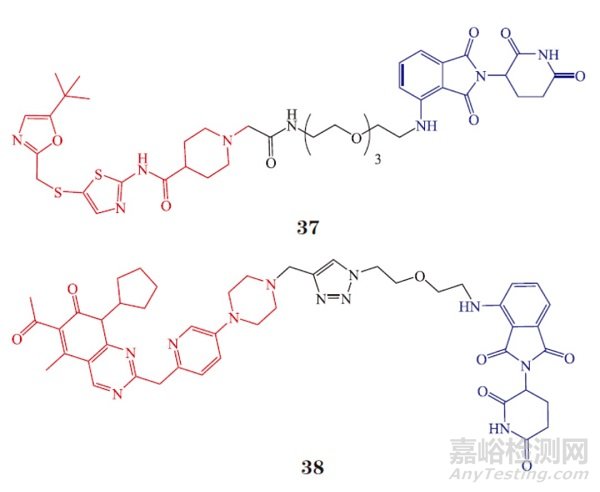
CDK 是真核生物中保守的丝氨酸 / 苏氨酸激酶家族,对细胞周期调节至关重要 [55]。CDK 家族共有 20 个亚型:CDK1、2、4 和 6 负责调控细胞周期,CDK7 ~ 13 负责调控基因转录;CDK14 ~ 20 作用机制尚不清楚,但广泛地调控各种细胞活动 [56]。CDK9 是转录延长的重要调节因子,是肿瘤治疗的一个有希望的靶点,特别是对于由转录失调引起的肿瘤 [57]。Nathanael S. Gray 课题组通过研究 SNS-032/CDK2 共晶结构(PDB : 5D1J)[58],设计合成了名为THAL-SNS-032 的小分子 PROTAC 化合物 [33]。THAL-SNS-032(37)保持了对 CDK1/CycB(IC50 =171 nmol ·L-1)、CDK2/CycC(IC50 = 62 nmol ·L-1)、CDK7/CycH/MNAT1(IC50 = 398 nmol · L-1)和 CDK9 CycT1(4 nmol · L-1)的泛抑制活性。更有趣的是, THAL-SNS-032 在 5 μmol · L-1 时可以仅降解 CDK9 而对其他 CDK 靶点几乎没有降解效果,说明通过靶蛋白与 E3 泛素连接酶之间的协同作用,PROTAC 分子可以在小分子抑制剂的基础上提高其选择性。最近,饶燏课题组通过系统地考察 PROTAC 连接链、连接链的成分、POI 配体以及亲和力,发现了代表性化合物 CP-10 [59]。CP-10(38)是基于首个CDK4/6 的双功能抑制剂 palbocilid 的 PROTAC,其仅对CDK6 表现出明显的降解作用(DC50 = 2.1 nmol · L-1)。该研究表明,通过引入 PROTAC 技术可以将泛抑制剂转变成选择性降解剂,从而提高选择性和靶向性。
3.1.3 可通过降解整个蛋白影响非激酶依赖型功能
在过去的几十年发展中,酶抑制策略已被证明是许多传统小分子药物开发的有效手段。但是对某些非激酶依赖活性的疾病难以发挥有效的药理活性。
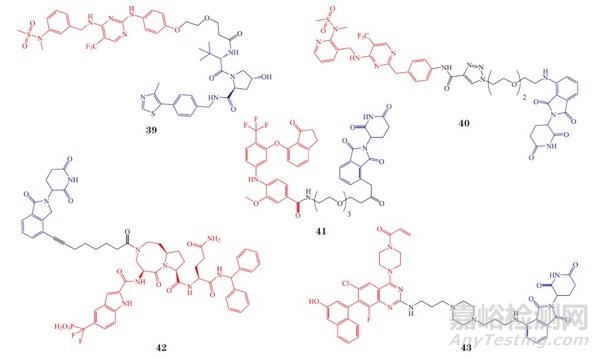
黏着斑激酶(FAK)是肿瘤入侵、转移的关键因子,同时也是多种信号蛋白的激酶和支架 [60]。尽管小分子 FAK 抑制剂在临床前和临床研究中取得一定进展 [61],但 FAK 支架作用介导的许多基本功能仍然是激酶抑制剂所无法抑制的 [61-62]。为克服激酶抑制剂难以靶向 FAK 的缺点,Craig M. Crews 课题组研制了高选择性、高活性的 FAK 降解化合物PROTAC-3(39,DC50 = 3.0 nmol·L-1,Dmax = 99%)[63]。PROTAC-3 在 FAK 激活以及 FAK 介导的细胞迁移和入侵方面均优于临床候选化合物 defactinib。其他作用于 FAK 的代表性小分子 PROTAC 还有 FC-11(40)[64] 和 BI-3663(41,DC50 = 27 nmol·L-1,Dmax = 95%)[65]。THAL-SNS-032(37)保持了对 CDK1/CycB(IC50=171nmol·L-1)、CDK2/CycC(IC50=62nmol·L-1)、CDK7/CycH/MNAT1(IC50 = 398 nmol · L-1)和 CDK9 CycT1(4 nmol · L-1)的泛抑制活性。更有趣的是, THAL-SNS-032 在 5 μmol · L-1 时可以仅降解 CDK9 而对其他 CDK 靶点几乎没有降解效果,说明通过靶蛋白与 E3 泛素连接酶之间的协同作用,PROTAC 分子可以在小分子抑制剂的基础上提高其选择性。最近,饶燏课题组通过系统地考察 PROTAC 连接链、连接链的成分、POI 配体以及亲和力,发现了代表性化合物 CP-10 [59]。CP-10(38)是基于首个CDK4/6 的双功能抑制剂 palbocilid 的 PROTAC,其仅对CDK6 表现出明显的降解作用(DC50 = 2.1 nmol · L-1)。该研究表明,通过引入 PROTAC 技术可以将泛抑制剂转变成选择性降解剂,从而提高选择性和靶向性。PROTAC 提供了同时阻断 FAK 激酶信号和支架能力的可能性。该研究证明了 PROTAC 具有拓展药物靶点空间和控制蛋白质功能特别是非激酶依赖型功能(kinase-independent function)方面的潜力,而这些作用对于传统的小分子抑制剂是不容易解决的。
3.1.4 有望降解“不可成药”的靶点
传统的小分子通过直接作用于靶蛋白表面具有一定深度的蛋白结合口袋(binding pocket)来调节蛋白功能。据统计, 人类超过 80% 的蛋白属于“不可成药”靶点 [66]。通常“不可成药”靶点蛋白表面平坦光滑,传统小分子难以寻找到有效的结合位点(binding site)。
信号转导与转录激活因子 3(STAT3)是细胞表面受体向细胞核传递信号的转录因子家族中的一员。STAT3 信号激活在细胞生长、分化、凋亡、代谢及抑制肿瘤免疫反应等过程中扮演极其重要的作用 [67]。STAT3 是人类肿瘤和其他疾病极具吸引力的治疗靶点。STAT3 结构特殊,缺乏传统小分子抑制剂可以直接作用的结合口袋,因而直接影响小分子抑制剂的研发。尽管在过去的几十年中有相关的直接抑制剂被报道,但效果不佳且缺乏特异性,目前临床上尚无有效药物 [68]。王少萌课题组通过基于结构的优化策略从先前报道的 STAT3 高亲和力拟肽化合物 CJ-887 出发,优化得到 SI-109。分析 SI-109 与STAT3 的共晶结构(PDB : 6NUQ)[69],设计合成了STAT3 降解剂 SD-36(42)(DC50 = 60 nmol ·L-1)[70]。该研究工作首次证明 PROTAC 技术可以将“不可成药”靶点转化为“可成药”(druggable)靶点,为后续 PROTAC 技术在“不可成药”靶点方面的应用奠定基础。
RAS 是癌症中最为常见的突变癌基因,也是肿瘤发生的重要驱动因素,约占所有癌症的 30%[71]。由于 RAS 在肿瘤发生、发展中的重要作用,其靶向治疗已成为抗肿瘤研究的一个热点。近年来,随着针对 K-RAS(G12C)突变蛋白的小分子药物深入开发,直接抑制 RAS 的抑制剂取得了一定成功。然而,这些抑制剂对 G12C 突变肿瘤的适用性仍非常有限。因此,RAS 基因过去一度被认为是“不可成药”靶点 [72]。目前已有学者报道了多种 E3 泛素连接酶如 Rabex-5[73]、亮氨酸拉链样转录调节子因子1(LZTR1)[74] 和 β-TrCP [75] 等参与 RAS 的泛素化降解。近日,Eric S. 和 Nathanael S. Gray 课题组利用 K-RAS 抑制剂通过连接链的优化得到基于 CRBN 的 PROTAC 分 子 XY-4-88(43)[76]。XY-4-88 可以诱导 GFP-KRASG12C 降 解。但 是,对内源性KRASG12C 不能起到降解作用。未来还需要进一步探讨 PROTAC 在降解内源性 KRAS 方面的研究,为进一步寻找更广泛有效的靶向 RAS 药物带来契机。
3.1.5 提供了一种新型的快速可逆的化学蛋白敲除方法
蛋白敲低策略是研究靶基因功能丧失后果的有效手段 [77]。从传统意义上来说,基因功能丧失的研究主要是通过RNA 干扰 [78]、基于重组基因的敲除、CRISPR-Cas9 [79] 等基因编辑技术来开展的。然而,这些策略难以实现快速可逆的蛋白敲低 [80]。传统的基因敲除与蛋白敲低技术实验周期长、费用高等缺点给研究者带来诸多挑战,尤其在大型非灵长类动物上。此外,许多基因的缺失将导致胚胎死亡,限制了相关科学研究 [81]。作为一种新型、快速、高效的蛋白敲低模型的方法,PROTAC 是现有遗传性工具的有效补充手段 [29, 82]。
3.2 PROTAC 面临的挑战及发展策略
3.2.1 打破“类药五规则”的规律
由于 PROTAC 分子是由三部分组成,不可避免地导致相对分子质量过大,且目前报道的 PROTAC 的相对分子质量基本在800 以上,均不符合 Lipinski“类药五规则”定律, 而该定律是小分子药物细胞通透性和生物利用度的一个重要指标 [83]。如何提高细胞摄取率、生物利用度以维持 PROTAC 在生物体内必要的暴露量,以及获得具有理想的物理化学性质的分子将是一大挑战。面对这一挑战,饶燏课题组系统地研究了 PROTAC 在小鼠、猪和恒河猴体内的效果 [29]。结果表明,体内实验中 PROTAC RC32 可以显著降低 FKBP12(DC50= 0.27 nmol · L-1)和 BTK 的浓度。虽然 PROTAC 相对分子质量大,细胞通透性差,但还是可以通过连接链的优化,以及靶蛋白配体的选择来克服的。目前对于PROTAC 透过细胞膜的机制还不清楚,后续还需要更多的理论和实践来支撑 PROTAC 的吸收、分布、代谢、排泄以及毒性研究。
3.2.2 继续攻克“不可成药”靶点
人体中多数药靶为“不可成药”靶点。如何攻克“不可成药”靶点, 为疾病的治疗提供多种选择是未来药物研发必须面临的考验。
目前报道的基于 PROTAC 技术的靶蛋白小分子降解剂靶向的多数靶点为可成药靶点,而“不可成药” 靶点甚少。未来还需要深入研究,获取足够的证据来佐证靶向“不可成药”靶点的事实。
3.2.3 建立科学的评价体系
由于 PROTAC 在体内能以亚化学剂量发挥催化循环作用,因此传统的药代动力学(PK)、药效动力学(PD)方法不能很好地评估 PROTAC 的 PK 和 PD 性质,针对 PROTAC 分子,目前尚无成熟的 PK 和 PD 评价体系,量效关系、时效关系的规律尚未完全掌握,蛋白降解所致的毒性尚未透彻了解。未来亟需建立合适的 PK 和 PD 评价体系。
3.2.4 完善 PROTAC 的理性药物设计
PROTAC 分子设计也是 PROTAC 技术面临的一大挑战。目前仅有少数科学家成功解析出 PROTAC 的三元复合物 [45, 84]。Ciulli 课题组解析了 PROTAC 分子 MZ1 与靶蛋白 BRD4、E3 泛素连接酶 VHL 的三元复合物晶体结构(PDB : 5T35)[45],发现 MZ1 在拉近靶蛋白和 E3 泛素连接酶的同时,还可诱导靶蛋白和 E3 泛素连接酶结合界面之间的相互作用,连接链的组成和长度都对这种协同作用有较大影响。这对后续PROTAC 的理性设计具有实际指导意义。未来还需要更细致地从结构上进一步阐明 PROTAC 的作用机制以及开展更多的构效关系研究,为 PROTAC 的分子设计提供更多的指导。
3.2.5 拓展 E3 泛素连接酶配体
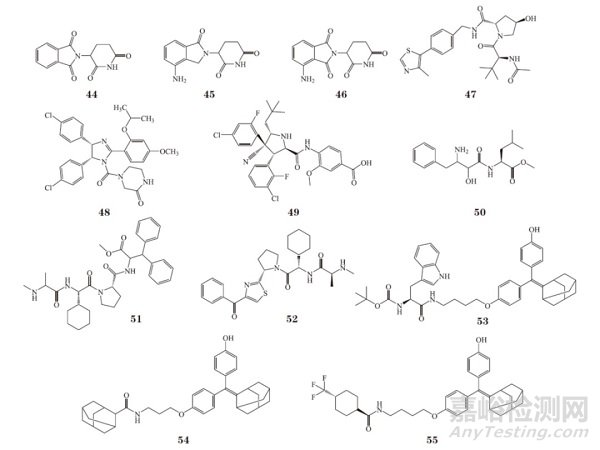
在蛋白酶体介导的蛋白降解过程中,E3 泛素连接酶是至关重要的组成部分。人体中已知的 E3 泛素连接酶有 600 多个,但迄今仅不到 1% 的 E3 泛素连接酶具有小分子配体 [85], 目前报道的 90% 以上的 PROTAC 小分子化合物均以最为常见的 E3 泛素连接酶为招募对象,例如CRBN、VHL、MDM2、cIAP1[86]。如何拓展可用于PROTAC 技术的 E3 泛素连接酶也是 PROTAC 所面临的挑战之一。未来能否寻找到在特定细胞或组织中具有特异性的 E3 泛素连接酶及其配体也是必须考虑的一大科学问题。常见 E3 泛素连接酶配体有沙利度胺(44)、来那度胺(45)、泊马度胺(46)、VHL 配体(47)、(-)-nutlin 3(48)、idasanutlin(49)、methyl bestatin(50)、MV1 配体(51)、cIAP 配体(52)、氨基酸选择性雌激素受体降解剂(SERD,53)、桥接三环 SERD(54)和单环 SERD(55)等。
3.2.6 其他
PROTAC 只有在形成稳定 的“靶蛋白-PROTAC-E3 泛素连接酶”三元复合物时才能高效特异性泛素化靶蛋白。然而,由于三元复合物的复杂体系难以捕获,目前研究 PROTAC 介导的三元配合物的方法还不多见,主要有时间分辨荧光能量转移法(TR-FRET)、AlphaLISA 法、表面等离子体共振法(SPR)和等温滴定量热法(ITC)[87-88]。虽然这些方法对三元复合物的形成的分析都有一定价值,但并不能完整概括出 POI 降解所需要的泛素- 蛋白酶体系统。目前大多数研究工作主要探讨目标蛋白与泛素连接酶各自配体的“二元复合物”稳定性。未来将深入考察“靶蛋白-PROTAC-E3 泛素连接酶” 三元复合物体系的稳定性。
PROTAC 中间 Linker 的设计也至关重要,线性脂肪链烃或醚结构有被氧化代谢的风险,大大减少了药物暴露浓度与时间,加快 PROTAC 分子排出生物体外。如何构建 PROTAC 中间连接链的长度与组成现在还没有规律的认识。
最后,PROTAC 分子的复杂性给化学家也带来了巨大的挑战。Soural 研究团队运用固相合成技术快速高效地合成了基于沙利度胺的 PROTAC 分子 [89]。未来亟待继续开拓高效、快捷、无污染的合成技术平台,从而为临床研究以及后续的规模化生产提供充足的原料。
总的来说,任何干预内源性蛋白质调节机制的药物都需要高度的设计水平和特异性来调控一系列的生物学事件,这是一个重大的挑战。相信通过学术界和制药工业界广大科研人员的共同努力,这些问题在不远的将来都可以得到满意的解决方案。
04、结语与展望
起初,Craig M. Crews 把 PROTAC 描述为“cute chemical curiosity”,可能仅仅是出于一种学术上的好奇心。如今,PROTAC 技术已成为新药研发的新策略,为疾病的治疗提供新方法,未来几年将是PROTAC 发展的关键时期, 越来越多的 PROTAC小分子将进入临床前和临床研究,进一步检验PROTAC 的治疗效果。PROTAC 面临的各种问题将被逐一解决,成为继小分子抑制剂、单克隆抗体之后的又一种重磅抗肿瘤手段。
参考文献
Sun X, Gao H, Yang Y, et al. PROTACs: great opportunities for academia and industry[J]. Signal Transduct Target Ther, 2019, 4(64): 64. Doi:10.1038/s41392-019-0101-6.
Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy[J]. Science, 2019, 366(6467): 818-822.
Komander D, Rape M. The ubiquitin code[J]. Annu Rev Biochem, 2012, 81: 203-229.
Neklesa T K, Winkler J D, Crews C M. Targeted protein degradation by PROTACs[J]. Pharmacol Ther, 2017, 174: 138-144.
Fisher S L, Phillips A J. Targeted protein degradation and the enzymology of degraders[J]. Curr Opin Chem Biol, 2018, 44: 47-55.
Bondeson D P, Mares A, Smith I E, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs[J]. Nat Chem Biol, 2015, 11(8): 611-617.
Adjei A A. What is the right dose? The elusive optimal biologic dose in phase I clinical trials[J]. J Clin Oncol, 2006, 24(25): 4054-4055.
Coleman K G, Crews C M. Proteolysis-targeting chimeras: harnessing the ubiquitin-proteasome system to induce degradation of specific target proteins[J]. Ann Rev Cancer Biol, 2018, 2(1): 41-58.
Sakamoto K M, Kim K B, Kumagai A, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation[J]. Proc Natl Acad Sci USA, 2001, 98(15): 8554-8559.
Sakamoto K M, Kim K B, Verma R, et al. Development of PROTACs to target cancer-promoting proteins for ubiquitination and degradation[J]. Mol Cell Proteomics, 2003, 2(12): 1350-1358.
Schneekloth J S, Jr., Fonseca F N, Koldobskiy M, et al. Chemical genetic control of protein levels: selective in vivo targeted degradation[J]. J Am Chem Soc, 2004, 126(12): 3748-3754.
Buckley D L, Van Molle I, Gareiss P C, et al. Targeting the Von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction[J]. J Am Chem Soc, 2012, 134(10): 4465-4468.
Han X, Wang C, Qin C, et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer[J]. J Med Chem, 2019, 62(2): 941-964.
Zhao Q, Ren C, Liu L, et al. Discovery of SIAIS178 as an effective BCR-ABL degrader by recruiting Von Hippel-Lindau (VHL) E3 ubiquitin ligase[J]. J Med Chem, 2019, 62(20): 9281-9298.
Burslem G M, Schultz A R, Bondeson D P, et al. Targeting BCR- ABL1 in chronic myeloid leukemia by PROTAC-mediated targeted protein degradation[J]. Cancer Res, 2019, 79(18): 4744-4753.
Chan K H, Zengerle M, Testa A, et al. Impact of target warhead and linkage vector on inducing protein degradation: comparison of bromodomain and extra-terminal (BET) degraders derived from triazolodiazepine (JQ1) and tetrahydroquinoline (I-BET726) BET inhibitor scaffolds[J]. J Med Chem, 2018, 61(2): 504-513.
Burslem G M, Smith B E, Lai A C, et al. The advantages of targeted protein degradation over inhibition: an RTK case study[J]. Cell Chem Biol, 2018, 25(1): 67-77.
Vollmer S, Cunoosamy D, Lv H, et al. Design, synthesis, and biological evaluation of MEK PROTACs[J]. J Med Chem, 2019, 63(1): 157-162.
Gechijian L N, Buckley D L, Lawlor M A, et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands[J]. Nat Chem Biol, 2018, 14(4): 405-412.
Schneekloth A R, Pucheault M, Tae H S, et al. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics[J]. Bioorg Med Chem Lett, 2008, 18(22): 5904- 5908.
Vassilev L T, Vu B T, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2[J]. Science, 2004, 303(5659): 844-848.
Hines J, Lartigue S, Dong H, et al. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53[J]. Cancer Res, 2019, 79(1): 251-262.
Zhao Q, Lan T, Su S, et al. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule[J]. Chem Commun (Camb), 2019, 55(3): 369-372.
Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity[J]. Science, 2010, 327(5971): 1345-1350.
Lopez-Girona A, Mendy D, Ito T, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide[J]. Leukemia, 2012, 26(11): 2326- 2335.
Sievers Q L, Petzold G, Bunker R D, et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN[J]. Science, 2018, 362(6414): eaat0572. Doi:10.1126/science. aat0572.
Fischer E S, Bohm K, Lydeard J R, et al. Structure of the DDB1- CRBN E3 ubiquitin ligase in complex with thalidomide[J]. Nature, 2014, 512(7512): 49-53.
Qin C, Hu Y, Zhou B, et al. Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression[J]. J Med Chem, 2018, 61(15): 6685-6704.
Sun X, Wang J, Yao X, et al. A chemical approach for global protein knockdown from mice to non-human primates[J]. Cell Discov, 2019, 5: 10. Doi:10.1038/s41421-018-0079-1.
Lai A C, Toure M, Hellerschmied D, et al. Modular PROTAC design for the degradation of oncogenic BCR-ABL[J]. Angew Chem Int Ed Engl, 2016, 55(2): 807-810.
Remillard D, Buckley D L, Paulk J, et al. Degradation of the BAF complex factor BRD9 by heterobifunctional ligands[J]. Angew Chem Int Ed Engl, 2017, 56(21): 5738-5743.
Schiedel M, Herp D, Hammelmann S, et al. Chemically induced degradation of Sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on Sirtuin rearranging ligands (SirReals)[J]. J Med Chem, 2018, 61(2): 482-491.
Olson C M, Jiang B, Erb M A, et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation[J]. Nat Chem Biol, 2018, 14(2): 163-170.
Huang H T, Dobrovolsky D, Paulk J, et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader[J]. Cell Chem Biol, 2018, 25(1): 88-99.
Buhimschi A D, Armstrong H A, Toure M, et al. Targeting the C481S ibrutinib-resistance mutation in Bruton's tyrosine kinase using PROTAC-mediated degradation[J]. Biochemistry, 2018, 57(26): 3564-3575.
Zhang C, Han X R, Yang X, et al. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK)[J]. Eur J Med Chem, 2018, 151: 304-314.
Rana S, Bendjennat M, Kour S, et al. Selective degradation of CDK6 by a palbociclib based PROTAC[J]. Bioorg Med Chem Lett, 2019, 29(11): 1375-1379.
Yang H, Lv W, He M, et al. Plasticity in designing PROTACs for selective and potent degradation of HDAC6[J]. Chem Commun (Camb), 2019, 55(98): 14848-14851.
Itoh Y, Ishikawa M, Naito M, et al. Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins[J]. J Am Chem Soc, 2010, 132(16): 5820-5826.
Okuhira K, Ohoka N, Sai K, et al. Specific degradation of CRABP-II via cIAP1-mediated ubiquitylation induced by hybrid molecules that crosslink cIAP1 and the target protein[J]. FEBS Lett, 2011, 585(8): 1147-1152.
Shibata N, Nagai K, Morita Y, et al. Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands[J]. J Med Chem, 2018, 61(2): 543-575.
Itoh Y, Ishikawa M, Naito M, et al. Protein knockdown using methyl bestatin-ligand hybrid molecules design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins[J]. J Am Chem Soc, 2010, 132(16): 5820-5826.
Ohoka N, Nagai K, Hattori T, et al. Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin-proteasome pathway[J]. Cell Death Dis, 2014, 5(11): e1513. Doi:10.1038/cddis.2014.471.
Shibata N, Shimokawa K, Nagai K, et al. Pharmacological difference between degrader and inhibitor against oncogenic BCR-ABL kinase[J]. Sci Rep, 2018, 8(1): 13549. Doi:10.1038/s41598-018- 31913-5.
Gadd M S, Testa A, Lucas X, et al. Structural basis of PROTAC cooperative recognition for selective protein degradation[J]. Nat Chem Biol, 2017, 13(5): 514-521.
Zou Y, Ma D, Wang Y. The PROTAC technology in drug development[J]. Cell Biochem Funct, 2019, 37(1): 21-30.
Gao H, Sun X, Rao Y. PROTAC technology: opportunities and challenges[J]. ACS Med Chem Lett, 2020, 11(3): 237-240.
Roskoski R, Jr. Properties of FDA-approved small molecule protein kinase inhibitors[J]. Pharmacol Res, 2019, 144: 19-50.
Sun X, Rao Y. PROTACs as potential therapeutic agents for cancer drug resistance[J]. Biochemistry, 2020, 59(3): 240-249.
Salami J, Alabi S, Willard R R, et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance[J]. Commun Biol, 2018, 1: 100. Doi:10.1038/s42003-018-0105-8.
Sun Y, Zhao X, Ding N, et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies[J]. Cell Res, 2018, 28(7): 779-781.
Sun Y, Ding N, Song Y, et al. Degradation of Bruton's tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas[J]. Leukemia, 2019, 33(8): 2105-2110.
De Wispelaere M, Du G, Donovan K A, et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations[J]. Nat Commun, 2019, 10(1): 3468. Doi:10.1038/s41467-019-11429-w.
Huggins D J, Sherman W, Tidor B. Rational approaches to improving selectivity in drug design[J]. J Med Chem, 2012, 55(4): 1424-1444.
Malumbres M. Cyclin-dependent kinases[J]. Genome Biol, 2014, 15(6): 122. Doi:10.1186/gb4184.
Sun T, Co N N, Wong N. PFTK1 interacts with cyclin Y to activate non-canonical Wnt signaling in hepatocellular carcinoma[J]. Biochem Biophys Res Commun, 2014, 449(1): 163-168.
Huang C H, Lujambio A, Zuber J, et al. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma[J]. Genes Dev, 2014, 28(16): 1800-1814.
Misra R N, Xiao H Y, Kim K S, et al. N-(cycloalkylamino)acyl- 2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5- [[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent[J]. J Med Chem, 2004, 47(7): 1719-1728.
Su S, Yang Z, Gao H, et al. Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders[J]. J Med Chem, 2019, 62(16): 7575-7582.
Lee B Y, Timpson P, Horvath L G, et al. FAK signaling in human cancer as a target for therapeutics[J]. Pharmacol Ther, 2015, 146: 132-149.
Ott G R, Cheng M, Learn K S, et al. Discovery of clinical candidate CEP-37440, a selective inhibitor of focal adhesion kinase (FAK) and anaplastic lymphoma kinase (ALK)[J]. J Med Chem, 2016, 59(16): 7478-7496.
Beraud C, Dormoy V, Danilin S, et al. Targeting FAK scaffold functions inhibits human renal cell carcinoma growth[J]. Int J Cancer, 2015, 137(7): 1549-1559.
Cromm P M, Samarasinghe K T G, Hines J, et al. Addressing Kinase- independent functions of FAK via PROTAC-mediated degradation[J]. J Am Chem Soc, 2018, 140(49): 17019-17026.
Gao H, Wu Y, Sun Y, et al. Design, synthesis, and evaluation of highly potent FAK-targeting PROTACs[J]. ACS Med Chem Lett, 2019, 11(10): 1855-1862.
Popow J, Arnhof H, Bader G, et al. Highly selective PTK2 proteolysis targeting chimeras to probe focal adhesion kinase scaffolding functions[J]. J Med Chem, 2019, 62(5): 2508-2520.
Santos R, Ursu O, Gaulton A, et al. A comprehensive map of molecular drug targets[J]. Nat Rev Drug Discov, 2017, 16(1): 19-34.
Johnson D E, O'Keefe R A, Grandis J R. Targeting the IL-6/JAK/ STAT3 signalling axis in cancer[J]. Nat Rev Clin Oncol, 2018, 15(4): 234-248.
Beebe J D, Liu J Y, Zhang J T. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we?[J]. Pharmacol Ther, 2018, 191: 74-91.
Bai L, Zhou H, Xu R, et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo[J]. Cancer Cell, 2019, 36(5): 498-511.
Zhou H, Bai L, Xu R, et al. Structure-based discovery of SD-36 as a potent, selective, and efficacious PROTAC degrader of STAT3 protein[J]. J Med Chem, 2019, 62(24): 11280-11300.
Spencer-Smith R, O'Bryan J P. Direct inhibition of RAS: quest for the Holy Grail?[J]. Semin Cancer Biol, 2019, 54: 138-148.
Khan I, Rhett J M, O'Bryan J P. Therapeutic targeting of RAS: new hope for drugging the "undruggable"[J]. Biochim Biophys Acta Mol Cell Res, 2019, 1867(2): 118570. Doi:10.1016/j.bbamcr.2019.118570.
Yan H, Jahanshahi M, Horvath E A, et al. Rabex-5 ubiquitin ligase activity restricts Ras signaling to establish pathway homeostasis in Drosophila[J]. Curr Biol, 2010, 20(15): 1378-1382.
Bigenzahn J W, Collu G M, Kartnig F, et al. LZTR1 is a regulator of RAS ubiquitination and signaling[J]. Science, 2018, 362(6419): 1171- 1177.
Jeong W J, Yoon J, Park J C, et al. Ras stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesis[J]. Sci Signal, 2012, 5(219): ra30. Doi:10.1126/ scisignal.2002242.
Zeng M, Xiong Y, Safaee N, et al. Exploring targeted degradation strategy for oncogenic KRAS(G12C) [J]. Cell Chem Biol, 2019, 27(1): 19-31.
Chaible L M, Corat M A, Abdelhay E, et al. Genetically modified animals for use in research and biotechnology[J]. Genet Mol Res, 2010, 9(3): 1469-1482.
Elbashir S M, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells[J]. Nature, 2001, 411(6836): 494-498.
Moon S B, Kim D Y, Ko J H, et al. Recent advances in the CRISPR genome editing tool set[J]. Exp Mol Med, 2019, 51(11): 130. Doi:10.1038/s12276-019-0339-7.
Chan A W. Progress and prospects for genetic modification of nonhuman primate models in biomedical research[J]. ILAR J, 2013, 54(2): 211-223.
Dhanjal J K, Radhakrishnan N, Sundar D. Identifying synthetic lethal targets using CRISPR/Cas9 system[J]. Methods, 2017, 131: 66-73.
Guo J, Liu J, Wei W. Degrading proteins in animals: "PROTAC"tion goes in vivo[J]. Cell Res, 2019, 29(3): 179-180.
Edmondson S D, Yang B, Fallan C. Proteolysis targeting chimeras (PROTACs) in 'beyond rule-of-five' chemical space: recent progress and future challenges[J]. Bioorg Med Chem Lett, 2019, 29(13): 1555-1564.
Nowak R P, DeAngelo S L, Buckley D, et al. Plasticity in binding confers selectivity in ligand-induced protein degradation[J]. Nat Chem Biol, 2018, 14(7): 706-714.
Konstantinidou M, Li J, Zhang B, et al. PROTACs- a game-changing technology[J]. Expert Opin Drug Discov, 2019, 14(12): 1255-1268.
Zhou Y, Xiao Y. Chemoproteomic-driven discovery of covalent PROTACs[J]. Biochemistry, 2019, 59(2): 128-129.
Hughes S J, Ciulli A. Molecular recognition of ternary complexes: a new dimension in the structure-guided design of chemical degraders[J]. Essays Biochem, 2017, 61(5): 505-516.
Roy M J, Winkler S, Hughes S J, et al. SPR-measured dissociation kinetics of PROTAC ternary complexes influence target degradation rate[J]. ACS Chem Biol, 2019, 14(3): 361-368.
Krajcovicova S, Jorda R, Hendrychova D, et al. Solid-phase synthesis for thalidomide-based proteolysis-targeting chimeras (PROTAC) [J]. Chem Commun (Camb), 2019, 55(7): 929-932.

来源:药学进展


