您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-09-13 16:17
背景
BET蛋白家族包含四个异构体:BDR2、BDR3、BDR4、BDET。每个BET蛋白均包含两个溴结构域——N末端BD1和C末端BD2。泛BET抑制剂同时作用于四个BET蛋白的两个溴结构域,其显著的抗癌和抗增殖活性已经得到充分证明。实际上,大量的的泛BET抑制剂已经进入临床I//II期用于癌症治疗。然而泛BET抑制剂也有众多的副作用被报道。
由于四个亚型之间高度同源性,发现高亚型选择性的BET抑制剂极具挑战,然而BD1和BD2之间的同源性是更低的(靠近乙酰化赖氨酸识别口袋的关键残基改变导致),因此可能实现BD2选择性。
到目前为止,尚未见高选择性的BD2抑制剂报道,RVX-208BD2对BD1的选择性大约30倍,GSK340选择性稍高(BRD4 BD1 Kd=1.1μM,BRD4 BD2 Kd=7nM)。ABBV-744为nM级的BD2抑制剂,正在临床前研究,具有290倍的选择性(BDR2/3/4, BD2 vs BD1)。
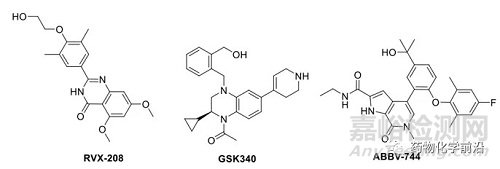
BD2选择性抑制剂用以理解BD2的体内生物学功能及为后续高选择性体内有效的BD2抑制剂的发现奠定基础。
苗头化合物
研究者首先对超过200万个化合物进行高通量筛选,为了筛选出仅仅结合BD2的小分子,将BD1乙酰赖氨酸结合位点的保守酪氨酸突变为丙氨酸(Y97A),使其结合位点失活,从而仅仅保留BD2具有结合活性(Y390A突变用于测试BD1活性)。最终得到1作为苗头化合物。
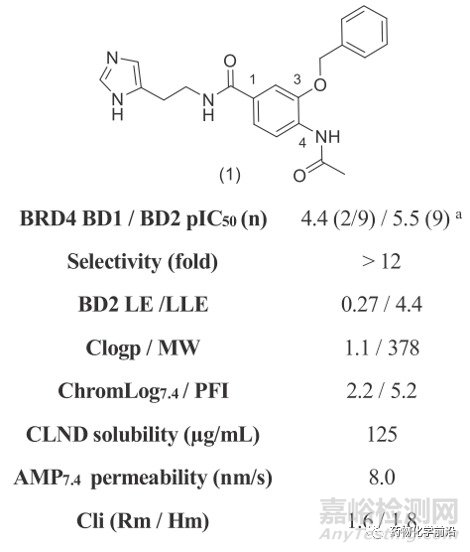
优化基础
为了进一步理解1的选择性,研究者获得了其与BRD4 BD1及BRD2 BD2结构域的共晶结构。化合物1包含与对乙酰氨基酚相似的乙酰化赖氨酸模拟母核,其结合模式与已经发表的扑热息痛在BRD2 BD1溴域的结合模式非常相似。N-乙酰基模仿组蛋白尾部乙酰化的赖氨酸,其羰基与保守的天冬氨酸(BRD4 BD1 Asn140)形成氢键,保守的水分子网络位于结合位点的底部。3位的末端苯环深入Ile146和Trp81-Pro82-Phe83形成的疏水口袋,1位的NH与Asn140形成水介导的氢键相互作用,此外,咪唑也与Asp96形成水介导的氢键网络。
1与BRD2 BD2的结合模式与BRD4 BD1类似,这类结合模式在泛BET抑制剂中很常见。小分子在BD2与BD1结合构象的不同在于咪唑的空间构象,咪唑临近的His433也存在两个旋转异构状态。在一个反式旋转异构体中,组氨酸侧链指向抑制剂,并允许两个水分子进入口袋界面,而在另一个旋转异构体状态下,侧链指向抑制剂并置换这些水分子。
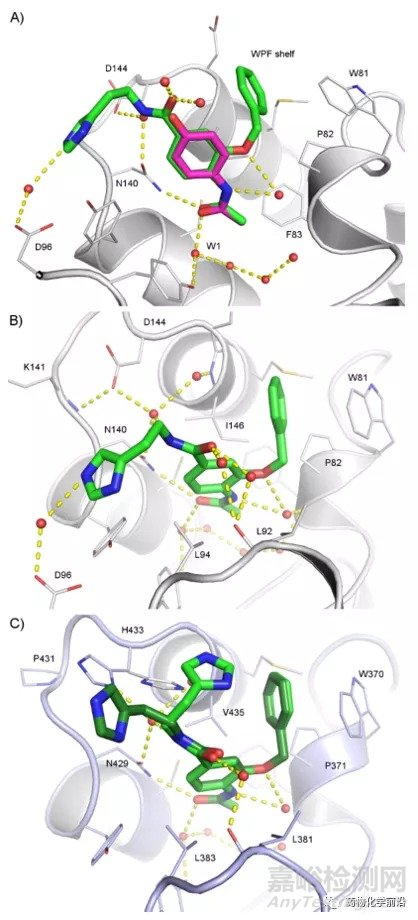
优化
因为碱性的氨基咪唑可能限制其透膜性,因此首先的SAR研究聚焦于酰胺咪唑部分,旨在提高其透膜能力。
其中,反式环己醇14提供了一个在效价、选择性、溶解度和渗透率方面具有良好平衡的化合物。
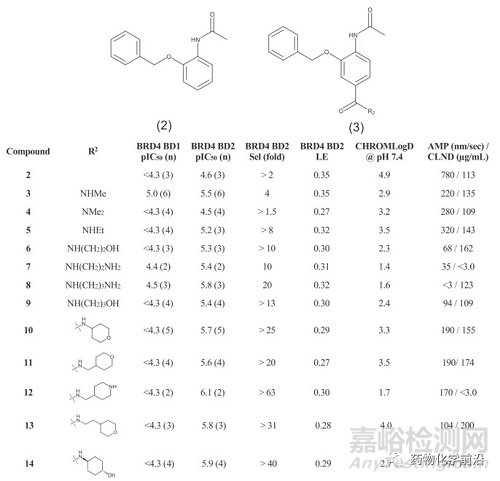
接下来研究者固定丙胺酰胺基团(早期研究中发现丙胺酰胺基团具有良好的效力、LE和选择性),对苄基苯部分进行SAR研究,结果显示在苄基的α位引入甲基,大大提高了活性和选择性,并保留了LE。
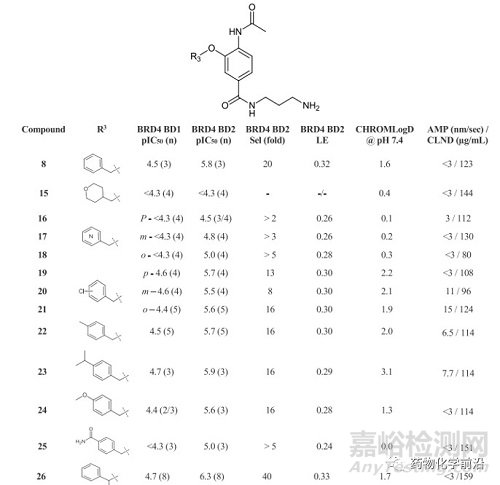
并且发现S对映体比相应的R对映体活性更高。
将两次优选的取代基组合得到化合物29,其显示了较好的BD2活性(将近100nM),杰出的选择性(250倍),并且保留了苗头化合物优秀的理化性质,在系列化合物中具有最佳的整体属性。
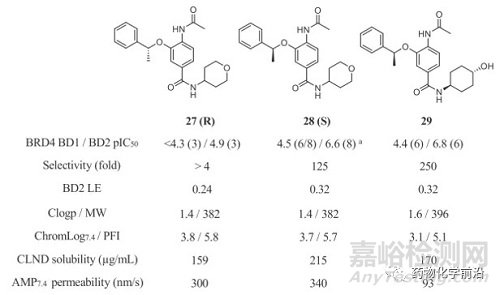
遗传毒性的避免
化合物29包含一个乙酰苯胺结构,在体内可能会由于脱乙酰作用形成一个诱变的氮鎓离子进而引发遗传毒性。代谢研究也确认46为脱乙酰的主要代谢产物。此外,化合物29在酸性条件下(2M HCl,24h)苄基将脱去,得到含有苯酚结构的47。
为了预测代谢中间体46及潜在的解离\代谢中间体47的的诱变性,应用eHOMO进行半经验计算。计算结果表明(结果>−0.320为Ames阳性)。降解/代谢中间体47具有最高的Ames阳性结果风险。进一步的Ames试验也证明了该计算结果。以上表明化合物26由于潜在的代谢/降解中间体47,推进临床具有巨大的风险。
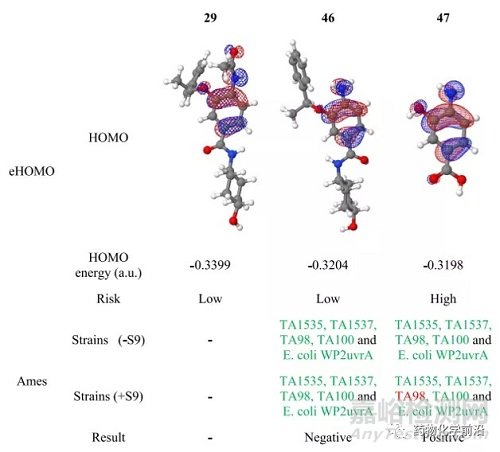
降低诱变风险的一个方法是通过在芳香母核引入吸电子基团降低苯胺的电子密度。
正如假设,在母核的各个位置引入F,得到了阴性的Ames检测结果。
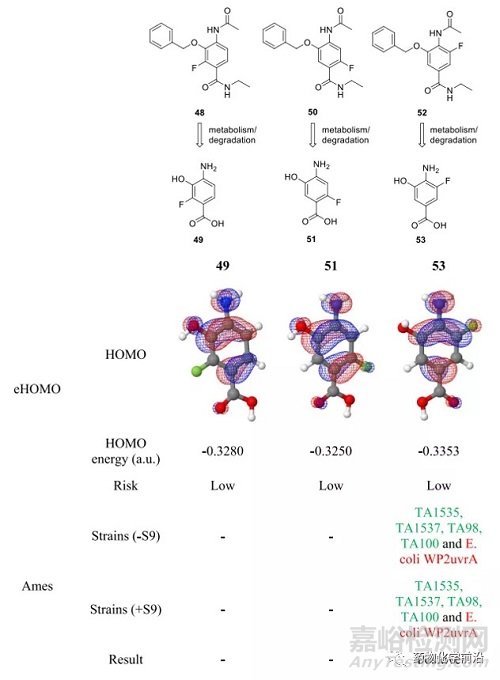
为了简化合成工作,将环己醇替换为乙基,在系列F取代的化合物中发现6F取代的化合物52是最佳的,BD2活性提高接近5倍。将优选的F取代和之前优化的环己醇组合,最终得到优选化合物59。
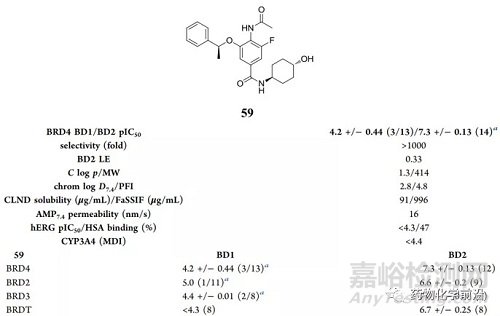
结果
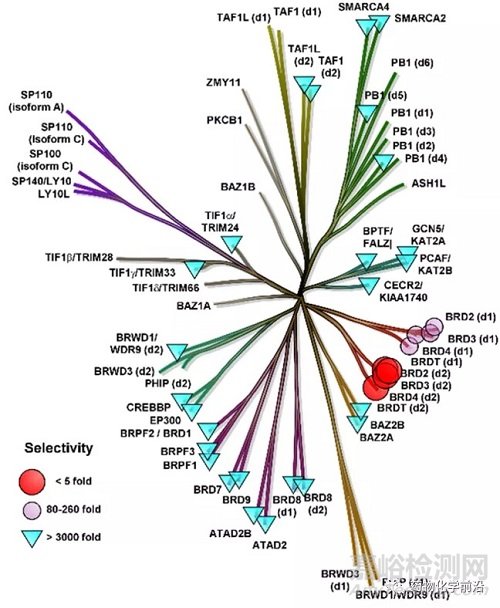
59对BET家族具有杰出的选择性,对非BET溴结构域无明显活性。
在LPS刺激的外周血单核细胞测试中,59抑制MCP-1(单核细胞趋化蛋白1,炎症相关)产生(pIC50=7.5),这不仅证明了59的细胞渗透性,同时也证明了BD2抑制剂也具有一定的抗炎潜力。
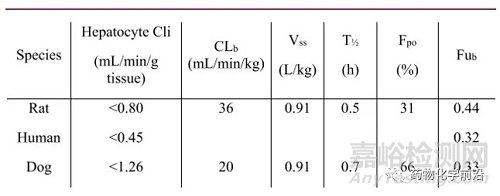
化合物59在大鼠和狗中显示了中等清除率,可接受的生物利用度,这些体内数据显示化合物59在动物模型中可作为一个合适的体内探针。
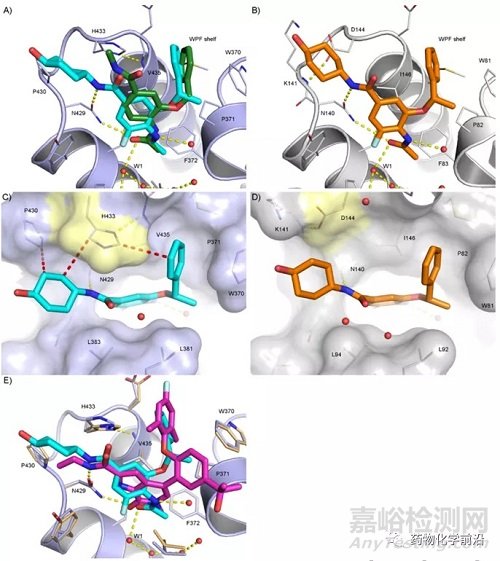
选择性的结构基础
59与BRD2 BD2及BRD4 BD1的共晶结构显示,该小分子的结合模式与1是相似的,4-乙酰氨基占据了乙酰化的赖氨酸口袋,并且与保守的W1水分子形成氢键相互作用。苄氧基占据了Val435及Trp370形成的口袋。
由于F原子与乙酰胺的静电排斥,化合物59乙酰胺部分相对于化合物1具有更多的扭转。这样的扭转迫使苄基更加靠近His433的侧链,进而形成变对面的相互作用,同时迫使酰胺的NH更靠近Asn429的羰基氧,增强氢键相互作用。而环己醇与Pro430及His433形成主要的疏水相互作用。
59与BRD4 BD1及BRD2 BD2的结合是相似的,选择性(BD2 vs BD1)的实现主要由于BD2中与His433(苄基/环己醇)及Pro430(苄基)更强的相互作用。
总结
泛BET抑制剂与BET蛋白家族的8个溴域具有同等的相互作用,并在许多体外表型分析和炎症或肿瘤学的临床前模型中显示出了显著的效果。大量的BET抑制剂已经推进临床,然而大多存在不良反应。为了更好的理解每个结构域对其效力的贡献并提高药物安全性,选择性的BET抑制剂亟待发展。
参考文献
Alex Preston, et al.Design and Synthesis of a Highly Selective and In Vivo-Capable Inhibitor of theSecond Bromodomain of the Bromodomain and Extra Terminal Domain Family ofProteins. J. Med. Chem. 2020, 63, 17, 9070–9092.

来源:药物化学前沿


