您当前的位置:检测资讯 > 检测案例
嘉峪检测网 2019-08-30 12:17
序言
仿制药上市都需要通过最后一道“终极考验”:BE实验。在仿制药研发过程会与参比制剂进行溶出曲线相似性对比后,才更有把握开展BE实验。当溶出曲线相似,BE不等效时,会有哪些问题方面的原因引起生物不等效呢?下面这篇文献描述了由于辅料不同的用量引起药物体内溶解后再沉淀不一致导致生物不等效。当我们遇到相似的药物时可以开展类似的研究,来增加BE实验通过的概率。
摘要
本研究旨在了解新开发的盐酸吡格列酮仿制药的生物等效性的原因,并改进其配方,使其与参比制剂(RLD)相当。在该临床研究中,尽管体外溶出度相似,但与RLD相比,新口服产品表现出较低的吡格列酮血浆浓度。吡格列酮溶解度为弱碱的强pH依赖性,表明吡格列酮溶解于胃中后会在小肠中沉淀。因此,进行体外实验以研究赋形剂对沉淀的吡格列酮粒度分布的影响,讨论了粒径对体内吸收的影响。来自RLD沉淀的吡格列酮显示出小颗粒(小于1μm)的峰,这在新开发产品的沉淀物中未观察到。作为赋形剂,羟丙基纤维素(HPC)影响沉淀的吡格列酮的粒径,在改良处方中HPC的量增加至与RLD中相同的水平。来自该改良处方的沉淀物显示出与RLD大致相同的粒度分布,并且在临床研究中成功地证明了生物等效性。总之,对于溶解度低的药物,除溶出试验外,这种沉淀药物粒度分布的分析可能有助于获得更好的体外-体内相关性,用于口服吸收和开发生物等效产品。
介绍
对于生物制药分类系统(BCS)中的II类药物(1,2)具有低溶解度和高渗透性,胃肠道(GI)中的药物溶解度和/或溶出速率被认为是限制口服吸收的主要因素(3)。各种技术用于制备这种难溶性药物,以通过提高溶解速率或溶解度来改善吸收,这些技术包括微粉化,成盐,共结晶,固体分散和使用增溶剂或基于脂质的制剂。为了确定这些制剂技术如何起作用,在进行体内研究之前,在体外观察每种制剂药物的溶出度。
在口服药物产品的生物等效性研究中,体外溶出试验也广泛用于确定活性成分的溶出度。对于普遍药物,要证明新开发的配方与参比制剂(RLD)的生物等效性,药物溶出度的相似性首先在体外进行,然后进行人体临床研究(4)。口服药物产品这一监管过程源于药物的药物口服胃肠道溶解曲线反映其吸收曲线。
药物吸收的体外-体内相关性(IVIVC)用于从其体外溶出试验中预测药物的口服吸收。然而,已经报道了各种研究,其中在体外药物溶出和体内吸收之间没有较好的相关性。药物吸收的IVIVC差可能源于这样的事实:溶出试验的体外条件不一定反映人体胃肠道的体内条件(5)。虽然使用模拟人体GI组成的生物相关溶出介质(例如,分别禁食或喂食状态模拟肠道,FaSSIF或FeSSIF)可显著提高可预测性(6–9),其他条件的差异,如流体体积和搅拌力,仍然是IVIVC相关差的原因。作为预测难溶性药物口服吸收的先进系统,开发了药物溶出与膜渗透的体外整合系统,以获得更好的IVIVC(10–18)盐,共结晶或无定形(固体分散)与其热力学平衡溶解度相比,可以以更高的浓度溶解药物。这种过饱和状态现在被认为是改善难溶性药物吸收的关键因素之一,并且已经进行了大量努力以在胃肠道中保持更高的浓度(19–22)。然而,一旦引发成核反应,过饱和会引起药物沉淀。沉淀的药物需要重新溶解在胃肠道中以被吸收到体循环中;否则,沉淀的药物不能吸收。如果这种沉淀和再溶解过程显著地影响药物吸收的程度和速率,则仅观察到药物溶出的体外试验可能不足以预测体内吸收。换句话说,应评估口服药物产品在胃肠道中的总体性能,不仅包括溶解,还包括沉淀和再溶解过程,以实现更好的IVIVC。
盐酸吡格列酮是口服抗糖尿病药,其被归类为BCSII类弱碱(pKa=5.8和6.8,logP=2.3)。盐酸吡格列酮的溶解度在很大程度上取决于pH值,显示为4.4mg/mL(pH1.2),0.042mg/mL(pH3.0),0.005mg/mL(pH4.0),0.0005mg/mL(pH5.0)和0.0003mg/mL(pH6.8)(23)。因此,口服给药并且在低pH条件下溶解在胃中,吡格列酮可能在小肠中沉淀或过饱和。在本报告中,根据我们对盐酸吡格列酮仿制药生物等效性研究的经验,研究了沉淀和再溶解过程对药物吸收的影响,重点是沉淀吡格列酮的粒度分布。
材料和方法
化学品和试剂
盐酸吡格列酮购自Tokuyama Corporation(Tokyo,Japan)。羟丙基纤维素(HPC)购自NipponSodaCo.,Ltd。(Tokyo,Japan)。低取代羟丙基纤维素(L-HPC)购自Shin-Etsu ChemicalCo.,Ltd(Tokyo,Japan)。D-甘露醇购自Mitsubishi Shoji Foodtech Co.,Ltd。(Tokyo,Japan)。甲醇,乙腈,乙酸和甲酸铵购自Wako Pure Chemical Industries,Ltd。(Osaka,Japan)。
药品
ACTOS®片剂30(ACT30)和ACTOS®OD片剂30(ACTOD30)购自Takeda Pharmaceutical Co.,Ltd(Osaka,Japan)。产品560T4SC704(SC704)和560T4SH906(SH906)是由Sawai Pharmaceutical Co.,Ltd(Osaka,Japan)开发的ACTOD30的仿制药。这些产品每片含有30毫克吡格列酮。
SC704中的赋形剂包括D-甘露醇,乙基纤维素,羟丙基甲基纤维素(HPMC),HPC,甘油三乙酸酯,L-HPC,阿斯巴甜,奇异果甜蛋白,交聚维酮,轻质无水硅酸和硬脂酰富马酸钠。SH906中的赋形剂包括D-甘露醇,HPC,L-HPC,阿斯巴甜,sodi-L-酒石酸盐,交聚维酮,轻质无水硅酸和硬脂酸镁。ACT30中的赋形剂包括乳糖水合物,羧甲基纤维素钙,HPC和硬脂酸镁。ACTOD30中的赋形剂包括D-甘露醇,乳糖水合物,微晶纤维素,羧甲基纤维素钙,HPC,阿斯巴甜,氯化钠,黄色氧化铁,交聚维酮和硬脂酸镁。
体外溶出度研究
使用日本药典中的桨法测定来自ACT30,ACTOD30,SC704和SH906的盐酸吡格列酮的溶出度(24)。简而言之,在溶出介质(每个容器900mL)中观察溶解药物浓度随时间变化过程,桨叶转速为50rpm,温度为37.0±0.5℃。使用pH1.2的JP1溶液(日本药典溶出试验液No.1)和pH6.8的JP2溶液(日本药典崩解试验液No.2)作为溶出介质。将样品通过0.45μm过滤器过滤,并通过高效液相色谱(HPLC)测定滤液。
生物等效性临床研究
生物等效性临床研究根据赫尔辛基宣言和良好临床实践指南进行。第一项研究由Doujin Memorial Meiwa医院(日本东京)进行,第二项研究由NITTAZUKA医疗福利中心福井综合医院(日本福井县)进行。这些研究得到了每个试验场所的机构审查委员会的批准。
将口服施用测试产品(SC704或SH906)后吡格列酮的血浆浓度与RLD(ACT30或ACTOD30)的血浆浓度进行比较。研究设计是双向,交叉,单剂量,禁食12小时和随机研究,7天清洗期。在他们被告知研究的目的,方案和风险后,在20或24名健康男性志愿者中进行了交叉研究。受试者在研究之前和期间至少7天未服用任何其他药物。在口服给予含有30mg吡格列酮的试验或RLD产品后,在0,0.5,1,1.5,2,3,4,6,8,12,24和34(或32)小时时取血样,将样品离心,分离血浆并储存在-20℃直至分析。用甲醇将血浆样品用1%稀释以沉淀蛋白质,并通过液相色谱串联质谱(LC/MS/MS)测定药物浓度。使用计算机软件BESTS(版本3.0.4,CAC Corporation,Tokyo,Japan)计算并统计比较药代动力学参数,其符合日本仿制药生物等效性测试的指导原则。如果测试与RLD的Cmax的几何均值比(测试/参考)在90%CI中间,AUCt值在80-125%之间则生物等效性
盐酸吡格列酮速释颗粒的制备
通过在塑料袋中摇动,加入16.5克盐酸吡格列酮,将24.5gD-甘露醇和7.5gL-HPC混合。随后,加入15.0g粘合溶液,并进行造粒。两种粘合剂分别为含有1%HPC和10%HPC水溶液(w/w)的溶液。用1%HPC和10%HPC水溶液(w/w)制备的颗粒分别缩写为“颗粒-1”和“颗粒-10”。将均匀造粒的产物在60℃的烘箱中干燥1小时。将得到的颗粒通过710μm筛筛分。
含沉淀颗粒的悬浮液的制备
通过以下步骤获得含有沉淀颗粒的悬浮液。将ACTOD30,SC704,SH906,颗粒-1或颗粒-10(各含有30mg吡格列酮)溶解于5mL JP1溶液中中,然后将1mL JP1加入200mL McIlvaine缓冲液(pH6.5)中。在混合后10或30分钟,将获得的悬浮液用于粒度测量。
在使用颗粒-1和颗粒-10的实验中,监测吡格列酮在McIlvaine缓冲液中溶解度随时间变化的过程。向McIlvaine缓冲液中加入1mL JP1后,在1,15,30,45,60,90,120和240分钟收集等分试样。立即通过0.45μm过滤器过滤样品,并通过LC/MS/MS测定滤液。
含沉淀颗粒溶液中粒度分布的测定
使用Mastersizer 2000(Malvern Instruments Ltd.,Worcestershire,UK),激光衍射粒度分析仪测量上述悬浮液中沉淀颗粒的尺寸分布。利用该设备,可以基于激光衍射技术测量0.01至2,000μm范围内的颗粒尺寸。关于粒度测量的优化条件,使用300rpm泵速而不进行超声处理以避免在测量期间沉淀颗粒的分解。通过平均一个样品的五次测量结果获得粒度分布曲线。
使用大鼠肠闭合环的原位吸收研究
所有动物实验均经由Setsunan大学的伦理审查委员会批准,并根据实验动物护理原则(NIH出版物No.85-23,1985年修订)进行。
为了评价HPC对吡格列酮肠吸收的影响,使用原位闭环法将含有从颗粒-1和颗粒-10得到的沉淀颗粒的悬浮液应用于大鼠肠道。雄性Wistar大鼠(SHIMIZU Laboratory Supplies Co.,ltd。,Kyoto,Japan),在实验前将体重200-250g(7-8周龄)禁食24小时,但可以自由饮水。打开腹腔后,通过插入硅胶管(3mm内径)在近端空肠处制作肠环(10cm长),然后通过缓慢注入盐水和空气除去肠内容物。使用相同的方法制备含有沉淀的药物颗粒的悬浮液。如上所述并储存10分钟之后将约0.5mL(2mL/kg体重)的悬浮液引入肠环中,并连接环的两端。在引入后5,30,60,90,120,180和240分钟从颈静脉采集血样。将样品离心,分离血浆并储存在-20℃直至分析。用甲醇将血浆样品稀释10%以沉淀蛋白质,然后通过LC/MS/MS分析。
药物浓度测定
使用LC/MS/MS系统测定样品中吡格列酮的浓度。LC系统由Shimadzu Corporation(Kyoto,Japan)的两个梯度泵(LC10Avp),自动取样器(SIL-HTC)和柱温箱(CTO10Avp)组成。分析柱是Cadenza CD-C18(30×2.0mm id,Imtakt Corporation Kyoto,Japan)。流动相由5mM甲酸铵溶液和乙腈和乙酸(1,000:1)的混合物组成。使用三重四极杆质谱仪API3000(AB SCIEX MA,USA)通过正离子模式的MS/MS检测实现定量。在多反应监测(MRM)模式中检测离子,其中在m/z357前体离子上的吡格列酮转变为m/z134产物离子。
结果
体外溶出度研究
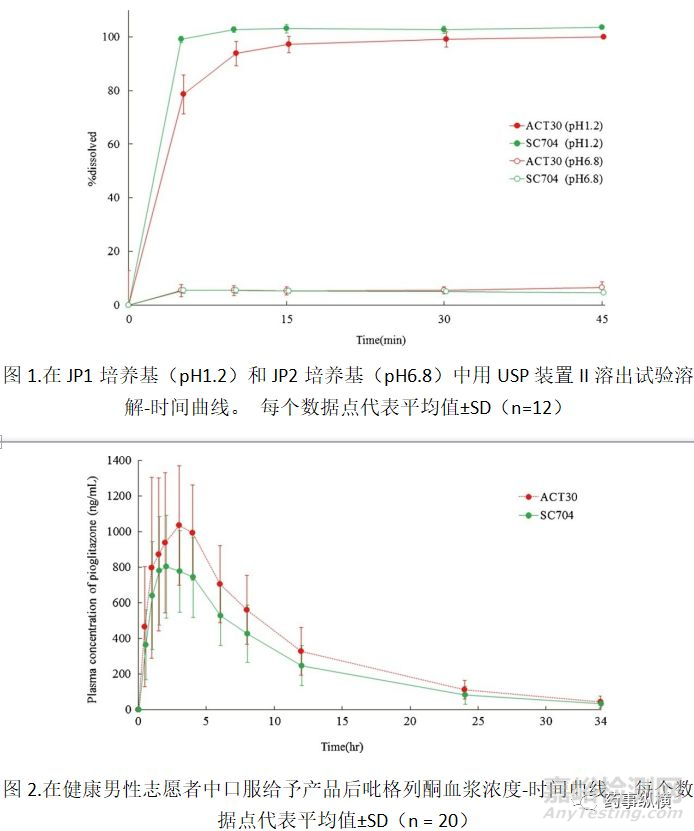
在JP1和JP2介质中评估来自测试产品(SC704)和RLD(ACT30)的盐酸吡格列酮的溶出度。结果如图1所示。.
盐酸吡格列酮的溶出度强烈依赖于其溶解度,我们在pH1.2和6.8时分别获得4.4和0.0003mg/mL的值(23)。在JP1介质中,盐酸吡格列酮在15分钟内几乎完全从两种产品中释放,因为盐酸吡格列酮的溶解度在pH1.2时足够高。当比较前10分钟期间的溶出速率时,SC704显示出与ACT30相比显著更高的速率。溶出速率的这种差异可能是因为SC704是口服崩解剂型,其被设计成在口腔中快速崩解。
相反,在JP2介质中,两种产品中溶解的盐酸吡格列酮的小于10%,因为其在pH6.8下的溶解度非常低。从这些体外溶出试验中,根据日本通用产品生物等效性研究指南,将这些产品评价为“相似”。
人体生物等效性研究
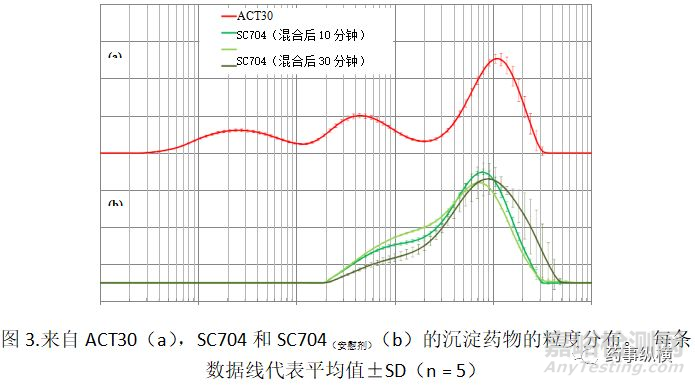
因为体外溶出试验显示两种产品的“相似”溶出度,所以进行临床研究以证实试验产品与RLD的“生物等效性”。图2显示口服SC704或ACT30后的健康志愿者吡格列酮血浆浓度-时间过程
在图2使用平均20名受试者,口服SC704在34小时内给予吡格列酮的血浆浓度低于ACT30的血浆浓度。结果,ACT30的AUCt和Cmax均高于SC704。根据日本指南,当AUCt和Cmax的几何均值比(测试/参考)的90%CI在预定的生物等效性接受限度(80-125%)内时,测试化合物和RLD可视为生物等效。然而,在该研究中,因为AUCt和Cmax的90%CI分别为68.3-83.6%和70.5-88.8%,SC704被认为与ACT30生物不等效。
沉淀物粒径的分布
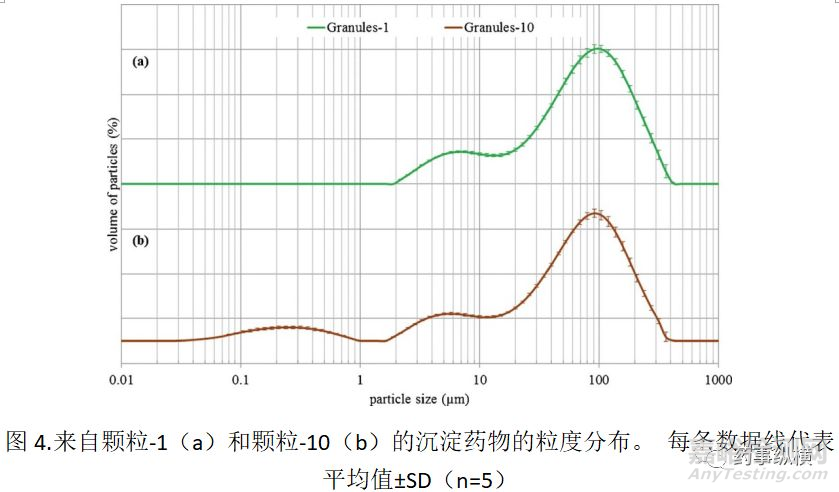
为了解释生物等效性研究中这种生物不等效性的原因,我们关注吡格列酮在胃肠道中的沉淀过程,因为溶出过程在体外溶出试验中被认为是相似的。制备含有沉淀颗粒的悬浮液,使用ACT30,SC704或SC704(安慰剂),后者不含吡格列酮。通过激光衍射观察这些悬浮液中粒径的分布,并示于图3中
如图3所示,在用ACT30和SC704制备的悬浮液之间观察到不同的粒度分布。在ACT30的情况下,在粒度分布的粒子中检测到三个峰,分别为约100,4和0.2μm。相比之下,SC704仅在约70μm峰和6μm处肩峰,但较小颗粒(小于1μm)的峰值消失。然而,由于这两种产品是不同的处方,包括不同的赋形剂和试剂,因此难以确定沉淀的吡格列酮的粒度分布的差异。颗粒尺寸分布如图3所示。包括不溶性成分的峰和它们的附聚物,例如崩解剂等。
HPC对沉淀吡格列酮粒径分布的影响
就这些处方的成分而言,我们假设HPC含量的差异可能会影响沉淀物的粒度分布。HPC尽管用量不同但是均在两种产品中作为粘合剂。从专利信息中估计ACT30中的HPC量为3.0mg(25)。在SC704配方中,将含有0.3mgHPC,0.3mgHPMC和6.0mg乙基纤维素的聚合物水分散体喷洒在33.1mg盐酸吡格列酮(含有30.0mg吡格列酮)来制备制粒前的包衣颗粒。然后,将包衣颗粒和用含有0.45mgHPC作为粘合剂的水溶液制粒。
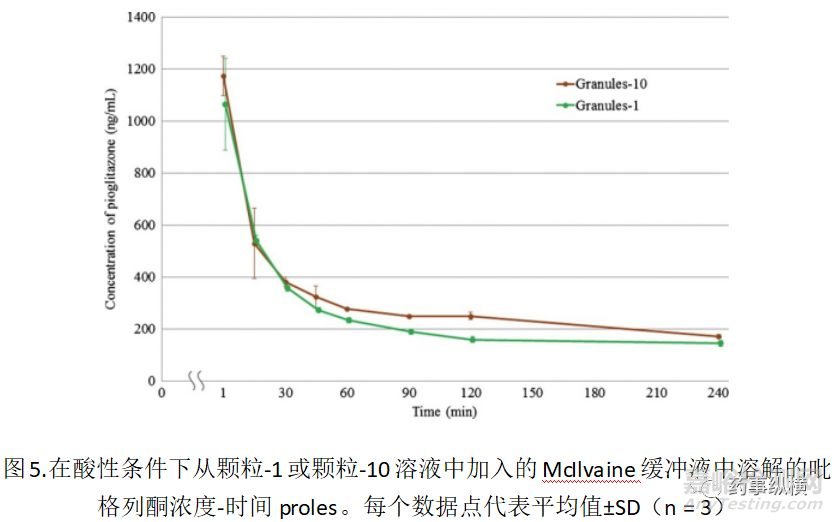
为了评估HPC对沉淀物粒度分布的影响,制备了两种类型的颗粒,颗粒-1和颗粒-10,其含有相同量的盐酸吡格列酮和赋形剂,HPC不同[对于颗粒-1,HPC/吡格列酮(w/w)=1/100(该比例基于SC704中盐酸吡格列酮上最佳的HPC量)和颗粒-10的10/100(该比率基于ACT30)专利信息)。
用颗粒-1和颗粒-10制备含有沉淀颗粒的悬浮液,并观察颗粒尺寸的分布。如图4所示。仅对于颗粒-10,在约0.2-0.3μm时观察到清晰的峰。颗粒-10的粒度分布的曲线类似于ACT30的粒度分布,如图3所示。虽然Granules-1产生了与SC704相似的类型。这些结果如图3 和4所示,清楚地表明,HPC量的差异导致沉淀物的粒度分布的差异。因为包含在颗粒中的HPC可溶于介质中,我们假设颗粒-10中观察到的直径小于1μm的小颗粒来自沉淀的吡格列酮。
将Granules-1或Granules-10溶解于5mL JP1介质中后,将1mL溶液加入200mL McIlvaine缓冲液(pH6.5)中,然后监测吡格列酮的溶解浓度以研究HPC在pH6.5下对吡格列酮的溶解浓度的作用(图5)。因为颗粒-1和-10的盐酸吡格列酮完全溶解于JP1介质中,所以吡格列酮在JP1介质中的溶解浓度为6mg/mL(30mg/5mL)。因此,McIlvaine缓冲液中溶解的吡格列酮的初始浓度理论上应该等于30μg/mL(稀释0.5%)。在第一次采样时间(稀释后1分钟),吡格列酮在McIlvaine缓冲液中的溶解浓度仅为1.2μg/mL,并逐渐降低至约240分钟时0.2μg/mL。HPC内容的差异(颗粒-1和-10)在吡格列酮的溶解浓度上没有引起很大差异。
HPC含量对吡格列酮肠道吸收的影响
使用原位大鼠肠闭环方法进行肠吸收研究以确定沉淀物的粒度分布的差异是否会影响吡格列酮的吸收。用颗粒-1和-10制备含有沉淀颗粒和附聚物的悬浮液,然后施加到大鼠肠环中以观察吡格列酮对血液循环的吸收。在将悬浮液应用于大鼠肠之前,通过HPLC测定溶解的吡格列酮的浓度。吡格列酮浓度未检测到显著差异(颗粒-10为680.3ng/mL,颗粒-1为704.9ng/mL)。
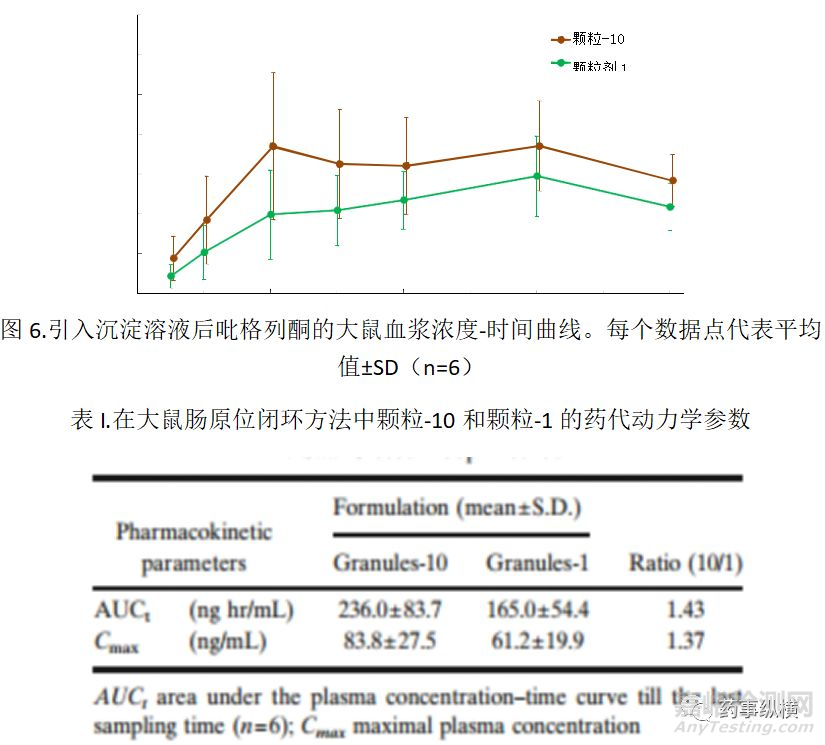
图6显示了在用含有沉淀物的悬浮液后吡格列酮的血浆浓度-时间过程。计算AUCt和Cmx并总结于表I中。虽然AUCt或Cmax未观察到显著差异,但平均而言,吡格列酮应用后的血浆浓度由颗粒-10制备的悬浮液高于颗粒-1制备的悬浮液。此外,当应用由颗粒-10制备的悬浮液时,血浆浓度在1小时迅速增加至Cmax。然而,在颗粒-1的情况下,血浆浓度逐渐增加直至施用悬浮液3小时。因为两种悬浮液中吡格列酮的溶解浓度大致相同,所以血浆浓度的差异可能是由于沉淀的吡格列酮的再溶解速率的差异引起的。图3所示的颗粒尺寸分布可能支持这个假设,因为沉淀的吡格列酮小颗粒(小于1μm峰)仅在由Granules-10制备的溶液中发现。因此,有理由认为这些小颗粒很快在肠道中重新溶解,被吸收到血液循环中。
含有高含量HPC的盐酸吡格列酮的新处方产品
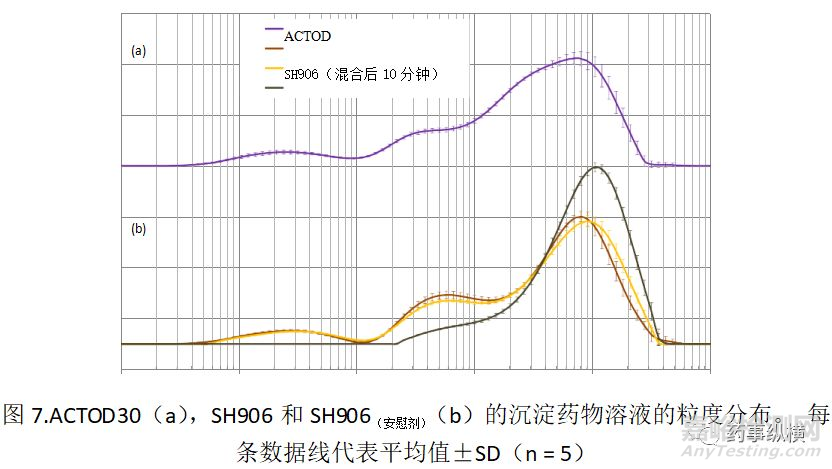
基于颗粒-1和-10的结果,开发了盐酸吡格列酮(SH906)的新处方。在SH906中,盐酸吡格列酮在制粒前没有包衣。用含有3.0mgHPC的水溶液作粘合剂,用于将盐酸吡格列酮与其它赋形剂一起制粒。另外,为了确定沉淀的吡格列酮的粒径,制备了含吡格列酮的SH906和不含吡格列酮的安慰剂SH906(安慰剂)。使用SH906,SH906(安慰剂)和RLD(ACTOD30)制备含有沉淀颗粒的悬浮液,以通过激光衍射观察粒度分布。
如图7所示,SH906显示出与ACTOD30类似的粒度分布,展示了约为0.2-0.3μm峰值。在制备悬浮液后10和30分钟观察到相同的曲线。相反,SH906(安慰剂)在小于1μm的范围内不产生峰。此外,在SC704或SC704(安慰剂)中未观察到较小的颗粒(小于1μm),因为在约0.2-0.3μm处的峰来自沉淀的吡格列酮,而不是来自其他赋形剂,包括HPC。该结果表明含有大量HPC的新处方(SH906)和RLD(ACTOD30)可能性具有生物等效性的。然后,进行生物等效性临床研究,结果如图8所示。
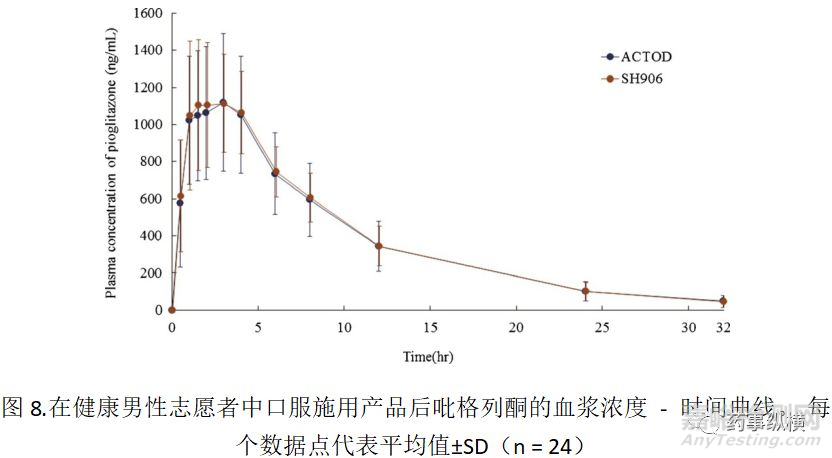
两种盐酸吡格列酮产品均表现出类似的吡格列酮血浆浓度;因此,在AUCt和Cmax中未发现差异。将AUCt和Cmax的几何均值比(测试/参考)90%CI分别为95.7-111.6%和96.6-113.6%,其在生物等效性的可接受限度内。
讨论
通常,为了证明新开发的口服药物产品与RLD的生物等效性,首先在体外测定溶出度的相似性,之后进行人体临床研究。然而,在这项研究中,尽管盐酸吡格列酮的新口服产品显示出与RLD相似的溶出度,但是口服给药后吡格列酮的血浆水平较低,研究未能证明新产品的生物等效性。
吡格列酮是一种水溶性差的药物,被归类为BCSII类。由于吡格列酮是弱碱,其盐酸盐在JP1介质中在酸性条件下迅速溶解,但在JP2中pH6.8时溶解度有限(图1)。根据这些体外溶出试验的结果,可以合理地认为,在口服给药后,盐酸吡格列酮完全溶解在胃中,然后吡格列酮在小肠中沉淀。沉淀的吡格列酮重新溶解在小肠中以被吸收到体循环中。
在酸性条件下,来自我们的处方(SC704)的盐酸吡格列酮的溶出速率快于来自RLD(ACT30)的溶出速率。临床生物等效性研究的结果如图2所示,其中SC704表现出较低的吡格列酮血浆浓度,在不假设赋形剂对药物沉淀影响情况下,随后的再溶解过程无法解释。因为SC704和ACT30组分中最显著的差异与HPC的量有关,我们关注HPC对吡格列酮吸收的影响。
水溶性聚合物,如HPMC和HPC,可维持难溶性药物的过饱和状态,并增强其口服吸收(20)。这些聚合物可防止药物分子在过饱和溶液中发生核反应(20–22,26)。如吡格列酮的情况,ACT30中较高量的HPC可能通过阻止小肠中的沉淀而导致吡格列酮的较高吸收。但是,如图5所示,HPC含量的差并异没有引起吡格列酮在pH6.5介质中溶解浓度(沉淀速率)随时间变化的显著差异。此外,在溶解的吡格列酮溶液(JP1)加入pH-6.5培养基后1分钟,吡格列酮的溶解浓度仅为1.2μg/mL。因为吡格列酮的初始溶解浓度理论上为30μg/mL,因此该结果表明大约96%的溶解吡格列酮是在将溶液加入pH-6.5培养基后1分钟内沉淀。因此,在吡格列酮的情况下,可以合理地认为HPC对过饱和的影响不是生物不等效性的主要原因。
含有用颗粒-1和-10制备沉淀颗粒的悬浮液显示出与分别对SC704和ACT30观察到的颗粒尺寸分布类似的颗粒分布(图3和图4)。在这两种情况下,包含较高量HPC的产品产生小于1μm的小颗粒,表明HPC防止溶液中颗粒的聚集。我们进行了另外的实验,其中将HPC预先加入到McIlvaine pH6.5缓冲溶液中;然后,将溶解SC704的JP1溶液加入缓冲液中。添加到缓冲液中的HPC总量与ACT30中含有的相同。结果,该实验对SC704的粒度分布曲线没有影响(数据未显示),表明添加到溶液中的HPC分子未能防止吡格列酮颗粒的聚集。从这些结果来看,提出以下作为HPC对沉淀的吡格列酮的粒径影响的可能机制。在沉淀过程中,包含在制剂中的HPC被认为在沉淀颗粒的表面上形成聚合物层。当颗粒彼此接触并且表面层重叠时,在颗粒之间产生重叠排斥能量。如果排斥能量大于范德瓦尔斯吸引力,则不会发生进一步聚集(27)。如ACT30和Granules-10情况。相反,如果HPC较少,则聚合物层变薄,范德瓦尔斯吸引力变得强于排斥能量,并且聚集,如SC704和颗粒-1所观察到颗粒情况的。通常,随着粒径减小,表面积增加,并且溶解速率增加。因为盐酸吡格列酮的口服生物利用度在动物中大于80%(28),如果溶出速率快,吡格列酮的肠道通透性足以完成快速吸收。换句话说,吡格列酮的口服吸收可能受溶解速率的限制,因此,在肠道中更快的溶解可促进吸收。如图6所示。用颗粒-10制备的包括沉淀颗粒的悬浮液显示出比用颗粒-1更高和更快的大鼠肠吸收。
最后,基于本研究获得的结果,我们通过增加HPC(SH906)的量来改善盐酸吡格列酮处方,并成功证明其与RLD(ACTOD30)的生物等效性。因为SH906的沉淀溶液含有小于1μm的小颗粒部分,并且颗粒尺寸分布的程度与ACTOD30几乎相同,我们认为沉淀药物的颗粒大小显著影响其吸收并改善盐酸吡格列酮制达到生物等效性。
结论
在该研究中,清楚地证明盐酸吡格列酮的口服制剂中HPC的量影响沉淀的吡格列酮的粒度分布。对于可能发生肠道沉淀的低溶解度药物,对沉淀药物的所得颗粒大小的分析可以提供对体内性能的了解。

来源:药事纵横


