您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2019-03-14 10:25

医疗器械UDI提前进入落地实施
这家医械公司已完成所有产品的UDI编码工作。
近日,威高医用制品公司完成了威高医疗器械唯一标识(以下简称“UDI”)所有的技术论证工作,所有产品的UDI编码工作已结束,建立了威高的UDI数据库,数据库与ERP系统和MES系统关联,实现了数据的共享与交互,至此,UDI工作提前进入到了实施阶段,威高医用制品公司在国内同行业率先实施了UDI管理。
国家药品监督管理局于2018年12月25日发布了医疗器械行业标准《医疗器械唯一标识基本要求》,实施日期为2020年1月1日,相关的法规《医疗器械唯一标识系统规则》和标准《医疗器械唯一标识系统术语与定义》《医疗器械唯一标识数据库基本数据集》也即将发布,标志着我国医疗器械行业全面实施医疗器械唯一标识已成定局。
据悉,UDI由三部分组成:UDI编码、UDI数据载体和UDI数据库。UDI编码采用符合GS1标准的Data Matrix二维码形式,每一个规格的产品都拥有一个全球唯一的标识码。该码作为产品唯一的“电子身份证”,可以在产品制造过程、仓储物流过程、医院耗材管理、国家不良事件监管、产品召回等产品生命周期全过程实现自动识别、精准追溯,为实现产品供应链的信息化管理奠定了基础,将对产品供应链效率的优化产生深远的影响。UDI数据库由产品标识与企业的营业执照、生产许可证、产品注册证和其他产品信息共同构成。通过UDI数据库,用户可以查询到该产品相关的所有信息。
威高医用制品公司UDI采用分阶段、分步实施推进。
在第一阶段,截止1月底,威高医用制品公司完成了产品外包装贴UDI标签工作,开发了包装上的UDI码自动赋码、标签联网打印系统以及针对销售端的出入库UDI扫码管理系统,实现扫码出入库,确保货物与单据的一致性,避免了人工输入带来的错误问题,并完成了系统内部调试和试用市场的现场调试工作,计划2019年3-5月份在市场推广使用。
第二阶段,威高医用制品公司计划在2019年8-12月份实现所有产品内外包装的UDI赋码工作,全面实施UDI管理。最终将根据管理需要开发UDI在仓库自动化管理中的应用和物流防串货等应用,并将UDI实施经验逐步推广至医用制品集团和全公司。
全面了解医疗器械UDI
2013年,国际医疗器械监管机构论坛(IMDRF)、美国FDA分别发布相关医疗器械唯一标识系统指南及法规。2014年,美国FDA率先对第三类医疗器械实施医疗器械唯一标识。
2017年5月,欧盟发布医疗器械法规,明确了实施医疗器械唯一标识的法规要求,日本、澳大利亚等国家也陆续开展相关工作,全球医疗器械唯一标识工作不断推进。
国家药品监督管理局于2018年2月26日发布《总局办公厅公开征求医疗器械唯一标识系统规则(征求意见稿)意见》。同年《医疗器械唯一标识通用要求》、《医疗器械唯一标识系统基础术语》两项标准审定会召开完成。这预示着NMPA UDI时代的到来。
今天就一起来聊一下UDI的相关要求。
1、什么是医疗器械追溯系统
医疗器械唯一标识系统,是指由医疗器械唯一标识、唯一标识数据载体和唯一标识数据库组成,共同构建的医疗器械统一识别系统。
解 释: 所谓的追溯系统,自然会包数据信息、信息载体及信息存储三部分。
2、医疗器械唯一标识
医疗器械唯一标识(Unique Device Identification,简称UDI),是医疗器械产品的电子身份证。
UDI 由DI 和PI 两部分组成,如下图:
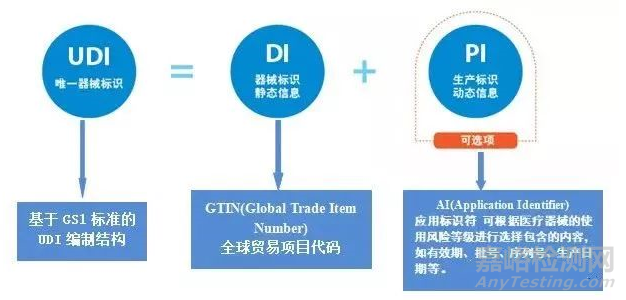
01 DI(Device Identifier)产品标识(或器械识别码)
产品标识(或器械识别码)是UDI 的固定和强制性部分,它包含贴标者(labeler)信息和产品型号。产品标识是识别医疗器械注册人或者备案人、医疗器械型号规格和包装的唯一码。
解释:DI一般组成:包装标识符 +厂商识别码+商品项目代码(一般为产品型号/规格)。通过产品标识就可以知道该产品相关的注册人或备案人是who?该产品的型号/规格是what ? 包装是what ?
02 PI(Production Identifier) 生产标识(或生产识别码)
生产标识(或生产识别码)是UDI 的可变和非强制性部分,根据监管和实际应用需求可包含医疗器械序列号、生产批号、生产日期、失效日期等。生产标识是识别医疗器械生产过程相关数据的代码。
解释:通过生产标识码可以了解该产品的生产信息,如产品什么时候生产的,产品啥时候失效等。
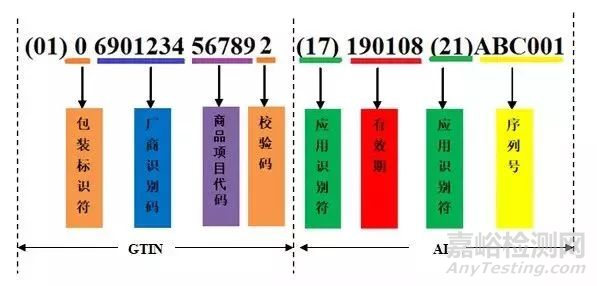
一个常见GS1编码构成的UDI例子:
|
项目 |
应用识别符 |
数据类型 |
最大 字符数 |
||
|
DI |
01 |
产品 识别符 |
包装标识符 |
数字 |
14 |
|
厂商识别码 |
数字 |
||||
|
商品项目代码 |
数字 |
||||
|
校验码 |
数字 |
||||
|
PI |
(17) |
有效期 |
数字 [YYMMDD] |
6 |
|
|
(11) |
生产日期 |
数字 [YYMMDD] |
6 |
||
|
(10) |
批号 |
字母+数字 |
20 |
||
|
(21) |
序列号 |
字母+数字 |
20 |
||
(1)厂商识别码由7-10位数字组成,中国物品编码中心负责分配和管理。厂商识别码的前3位为前缀码,国际物品编码协会已分配给中国物品编码中心的前缀为690-699。
(2)商品项目代码由厂商识别码所有人(即商品条码系统成员)依据有关国家标准自行分配。即企业依据相关要求及产品种类,对产品编制代码,一般是按规格/型号来编制。
(3) 校验码由标准算法得出。
(4) 包装标识符, 对于 GTIN-14代码的第1位数字为包装指示符,用于指示储运包装商品的不同包装级别,取值范围为1~9。但之前小编操作过国内UDI, 第一位都是固定的 0,条码中心只要求企业编辑商品项目代码备案。
(5)PI生产标识,为选择项,由企业根据医疗器械使用风险和监管追溯要求来确定其中的内容。
03 UDI的编制
医疗器械注册人或者备案人应当按照医疗器械唯一标识编制标准创建、维护医疗器械唯一标识。医疗器械唯一标识编制标准可包括国家食品药品监督管理总局认可的发码机构或者国家食品药品监督管理总局制定的相关标准。
解释:UDI不是随意编制的,医疗器械产品风险和监管追溯要求的不同,其器械的UDI也随之不同。UDI可单独由DI单独标识,也可由DI加PI联合使用标识。通常的做法是发码机构给医疗器械制造商分配唯一性的前缀,制造商在此基础上根据发码机构的标准给医疗器械产品分配完整的医疗器械唯一标识。
常见的UDI编制方式如下:
1)标识到规格型号
UDI仅由DI标识,可追溯到某公司某一型号/规格的医疗器械。
2)标识到批次
UDI由DI 联合PI中的生产日期/有效期、批号实现。可追溯到XX企业XX型号/规格医疗器械XX个批次。可用一维条码。
3)标识到单品。
UDI由DI 联合PI中的生产日期/有效期、序列号实现。可追溯到XX企业XX型号/规格XX个医疗器械。可用一维条码。
对于医疗器械来讲,大部分器械一般都要求追溯到批次,对于高风险的产品,如心脏起搏器需要追溯到单个产品。
04 UDI的基本原则
4.1唯一性:医疗器械唯一标识应当与医疗器械识别要求相一致。
解释:即对于相同特征的医疗器械,唯一标识的唯一性应当指向单个规格型号产品;对于采用序列号生产控制的医疗器械,唯一标识的唯一性应当指向单个产品;而对于按照批次生产控制的产品,唯一标识的唯一性指向同批次产品。此外对于唯一标识还要求即使该产品停止销售了,该产品的标识也不能不得用于其他医疗器械。但如若重新销售、使用时,可使用原产品标识。
4.2 稳定性:医疗器械唯一标识应当与产品基本特征相关,若产品的基本特征未变化,产品标识应当保持不变。
解释:在产品没有发生变化的条件下,UDI不能随便更改,不能由着心情,今天这样编制一个,明天来了个新leader看着不顺眼再换一个。因为后期UDI是要求产品注册时一并提供到NMPA。这里猜想,后续产品延续注册。
4.3可扩展性:医疗器械唯一标识应当与监管要求和实际应用不断发展相适应。
解释:这是对UDI编制标准制定者提出的要求,要求标准的制定要考到远期变化。
3、唯一标识数据载体
医疗器械唯一标识数据载体是存储和(或)传输医疗器械唯一标识的数据媒介。
要求如下:
01 载体可采用一维码、二维码或者射频标签等形式。鼓励采用先进的数据载体技术。
解释:目前仍以一维码为主。
02 采用一维码时,可将产品标识和生产标识串联,也可多行并联。采用射频标签时,应当同时具备一维码或二维码。
解释:目前基本都采用产品标识和生产标识并联的一维码方式。
03 数据载体需要标识在上市的医疗器械各级销售单元的包装或者医疗器械产品上,并确保在医疗器械经营和使用期间唯一标识数据载体牢固、清晰、可读。
4、唯一数据库
01 医疗器械唯一标识数据库定义:
数据库包含医疗器械的产品标识及相关数据。国家食品药品监督管理总局制定医疗器械唯一标识数据相关标准及规范,组织建立医疗器械唯一标识数据库,供公众查询。
解释:NMPA负责建立一个数据库,供公众查询,就跟医疗器械产品数据库一样。
02 唯一标识数据的提交要求:
注册人或者备案人应当在申请医疗器械注册或办理备案时,在注册/备案管理系统中提供申报产品的产品标识。
注册人或者备案人应当在其产品获准注册、取得备案或者变更后30个工作日内,将产品标识及相关数据上传至医疗器械唯一标识数据库。
解释:这里明确指出:产品注册/备案时需同时提交申报产品的产品标识且获得注册证或取得备案后录入标识数据库。这就意味着产品的UDI是需要备案的,就跟型号规格一样,不能随便更改。
5、UDI相关指南、标准及资料
01 总局办公厅公开征求医疗器械唯一标识系统规则(征求意见稿)意见
02 关于征求《医疗器械唯一标识系统 术语和定义》医疗器械行业标准意见的通知
03 关于征求《医疗器械唯一标识 通用要求》医疗器械行业标准意见的通知
04 基于GS1标准的五一器械标识(UDI)编制规范实现医疗器械可追溯
05 FDA UDI 实施规定以及如何申请唯一器械标识码
6、UDI的申请
根据之前申请的流程基本为:向中国物品编码中心申请厂商识别码,审批后,购买条码及软件。同时条码中心进行商品项目代码备案。企业最终会拿到医疗行业的UDI。具体可与中国物品编码中心在各地的分中心联系咨询。
7、注意事项
UDI的申请流程并不复杂,最大的问题是企业内部需要对DI中商品项目代码做好内部的规定。涉及到包装的还需要对不同包装做好规定。这就跟ERP系统建立时物料编码一样。不能随便编制一个,要考虑到系统性、长远性。目前UDI非强制性,仅仅是为配合医疗使用机构物流的方便而建立。但UDI强制性实施后,以后产品注册的时候UDI是需要同时备案的,这就要求在建立UDI时更需要谨慎。




来源:AnyTesting


