基于2021年12月,CFDI发布的《药品注册核查工作程序(试行)》等5个文件要求,药品注册核查目的主要可概括为三个:①数据可靠性的核实和/或实地确证、②核实申报资料的真实性和一致性、③核实商业化的生产条件。包括研制现场核查和生产现场核查,以及必要时的延伸检查活动。
研制现场核查是通过对药品研制合规性、数据可靠性进行检查,对药品注册申请的研制情况进行核实,对原始记录和数据进行审查,确认申报资料真实性、一致性的过程。研制现场核查的起点一般情况下为:确证性临床试验、生物等效性研究等药物临床试验相关批次,豁免药物临床试验的,以进行质量对比研究的相关批次为起点;未进行质量对比研究的,以工艺处方基本确定后的批次为起点。终点为:商业规模生产工艺验证批次前。包括药学研制现场核查、药理毒理学研究现场核查和药物临床试验现场核查等;重点为:临床试验批次、技术转移批次、稳定性试验批次等影响药品质量评价的关键批次。必要时,可前溯至研究立项、处方筛选、工艺优化等研究内容。
药学研制现场核查主要是对药学研制情况,包括药学处方与工艺研究、样品试制、质量控制研究及稳定性研究等研制工作的原始数据、记录和现场进行的核查。
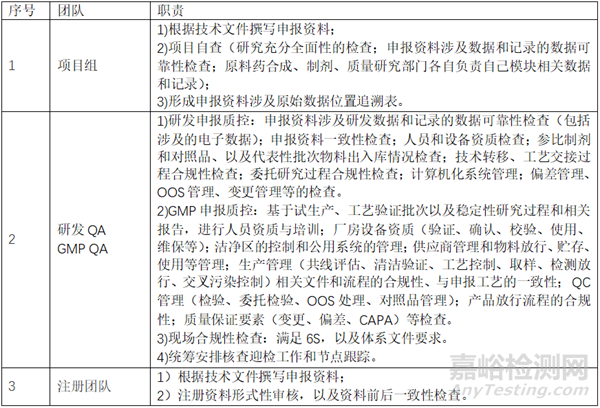
药品注册核查根据品种特性、被核查单位特点和风险、审评过程意见、有因检查的原因等,确定核查内容;各个公司职责划分也不相同。本文就常规原料药登记和制剂申报触发的注册现场核查,药学研制现场,研发团队负责项目开发和分析方法验证,GMP团队负责试生产、工艺验证、稳定性研究的情况,相关迎检准备工作和注意事项等介绍一二,以供大家参考。
一、 工作职责划分
建议在稳定性3个月左右启动项目自查工作,项目组完成自查后,稳定性4个月左右启动QA的申报质控。
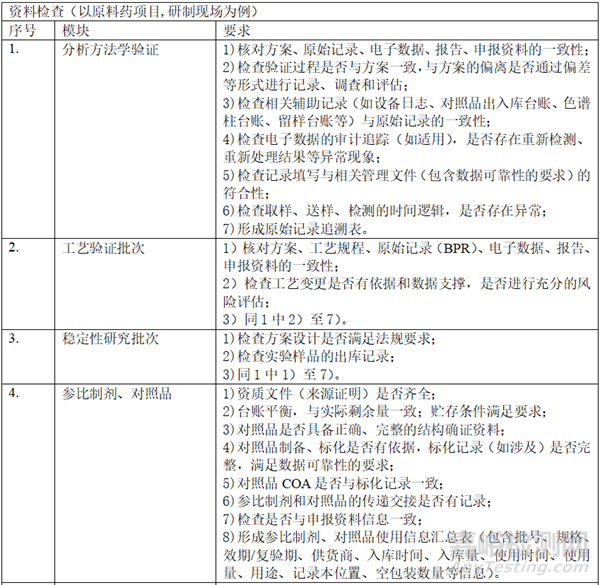
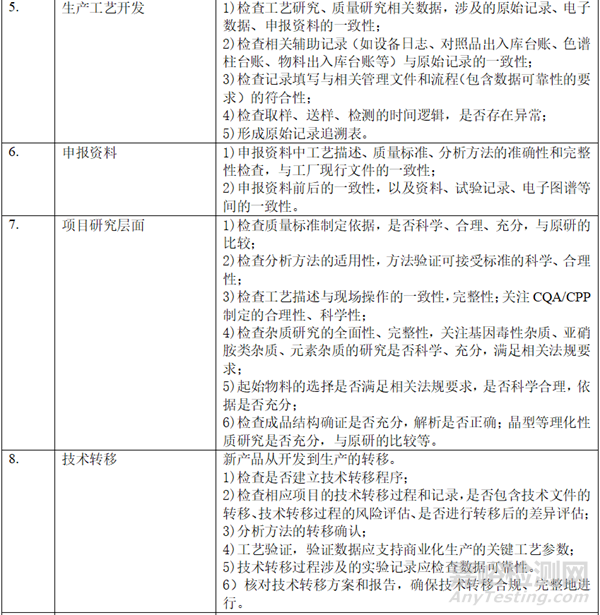
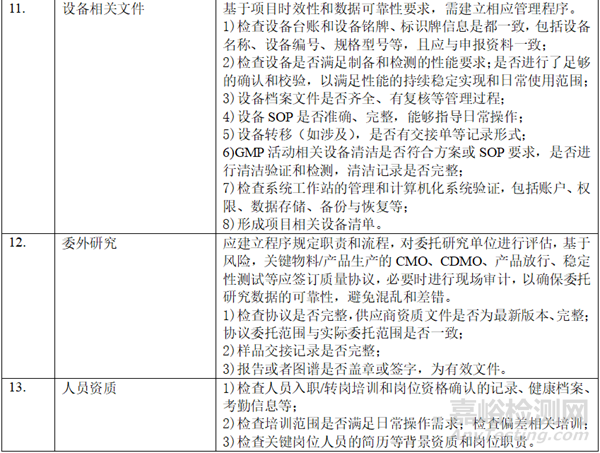
二、 资料准备
在自查的过程中,形成以下表单:样品试制清单(整体概况,批次、数量、时间、用途、剩余量)、研发和生产批次汇总信息表(代表性批次)、参比制剂使用信息梳理表(批次、资质、出入库数量和时间、用途等)、对照品清单(批次、资质、出入库数量和时间、用途等)、项目相关研究设备清单、项目相关检验设备清单、稳定性研究清单、研究人员清单、委托研究清单。
三、 现场准备
按照6S要求进行准备和自查,需要注意控制混淆、差错、污染、交叉污染的风险;关注物料标识、设备标识、环境控制(如涉及)、人员防护装备(实验服、手套、口罩等)的穿戴、人员操作和记录的及时性等。
核查期间,每天上班之前,可进行部门间的互查。
四、 人员准备
需要就文件传递、问题回答、现场介绍进行模拟演练,确定相关问题的回答人员,避免抢答、紧张、职责不清等引起混乱,并确保文件提供的准确性和及时性。