欧盟发布 MDCG 2023-7
2023年12月21日,欧盟发布了“MDCG 2023-7 - Guidance on exemptions from the requirements to perform clinical investigations pursuant to Article 61(4)-(6) MDR”关于根据 MDR 第61(4)-(6)条豁免临床研究要求的指南。
本指南旨在澄清将投放欧洲市场的植入式和III类医疗器械的临床研究要求豁免以及与等同性证明的相关条件。它还提供了与根据附录XIV第3节证明“足够程度的数据访问”相关的例子和考虑因素。
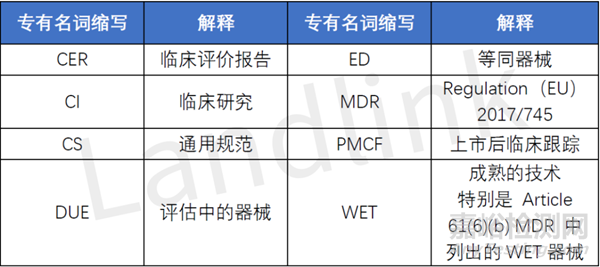
本文将涉及到诸多专有名词的缩写,具体如下:
1、植入式及III类器械豁免临床研究的要求
根据 Article 61(4) MDR 规定,应对植入式和III类器械进行临床研究,除非满足以下几种情况:
需要注意的是,这四个免于进行临床研究的情况相互独立。这意味着,除非直接引用,否则其中一种情况中概述的条件不适用于其他情况。
在情况1至3中的任何一种情况下,制造商可以在不签订合同的情况下,在DUE的临床评价中使用另一制造商的ED生成的临床数据。这方面的唯一要求是满足附录XIV第3节中描述的等同性标准。
ED数据的使用可以使制造商证明其临床评价是基于“足够的临床数据”。然而,只有满足上述情况中豁免条件的医疗器械,制造商才能免于进行临床调查。
2、等同性声明所需的"数据获取的充分程度"
除了概述证明等同性所需考虑的技术、生物学和临床特征外,附录XIV第3节MDR还要求制造商“[…] 有充分的条件获取具有等同性的相关器械上数据,以证明其等同性声明”。
如上节表格所述,证明“数据获取的充分程度”并不需要在所有情况下签订合同。合同仅适用于 Article 61(5) MDR 所述的豁免情况。还应注意的是,附录XIV第3节专门提到了证明等同性声明所需的数据:即,要求有足够的获取途径来确定评估等同性所依据的临床、技术和生物特征,而不是获得完整的技术文件。
Article 61(5) MDR所述的两家制造商签订的合同被推定为能够提供了证明等同性所需的全部数据。然而,在不需要合同的情况下,其他获取数据的手段也足以支持等同性证明,例如前文提到的情况1-3及情况4的特殊子集。
在情况4中,MDR不禁止在DUE的临床评价中使用来自多个制造商ED的临床数据。在这种情况下,只需要与一家制造商签订合同,就可以豁免临床调查,并在没有合同的情况下使用其他制造商器械的数据(证明其等同性)来补充临床评价中的数据。但DUE的制造商必须在CER中陈述他们充分获取数据级别的理由,并且必须得到公告机构的认可。
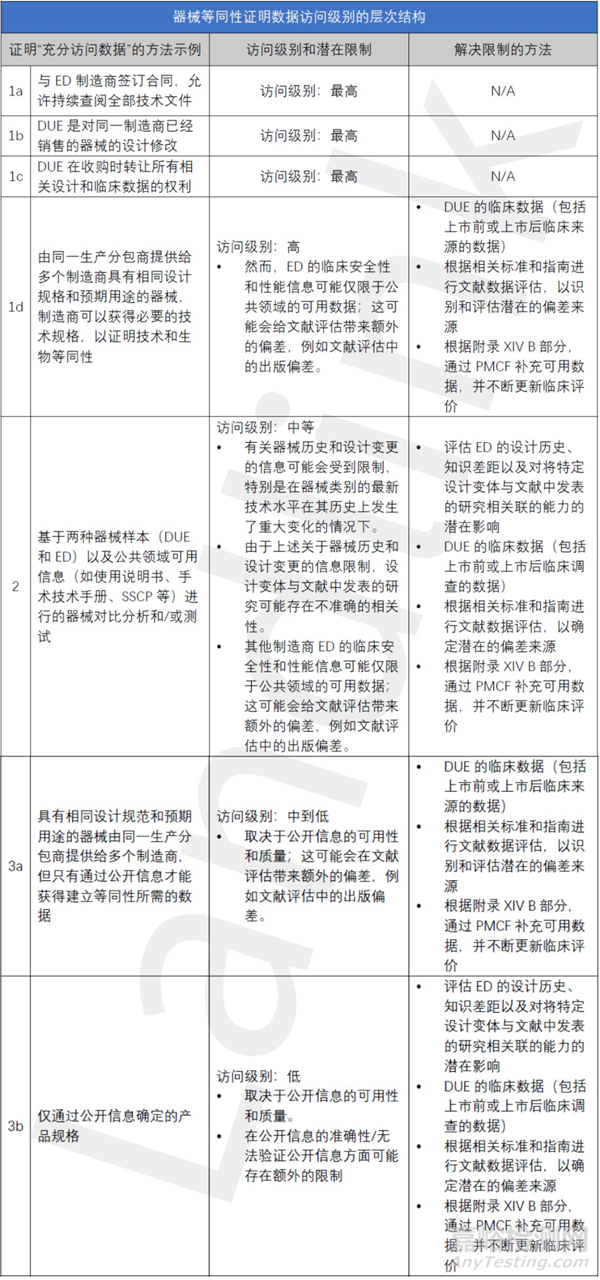
下表提供了一些与证明等同性相关的数据获取方式的示例,并提出了与该数据获取级别相关的等级结构。它还指出了这些方法和手段的潜在局限性,可以通过这些方法和途径来解决这些局限性,但此表仅用于说明目的,并非涵盖所有情况或是规定性的。