近日,国家药监局核查中心更新了一则关于中间产品存放时限相关的问答,提问问题涉及中间产品存放时限验证的具体批次如何选择?若产品工艺简单、物料性质稳定、不易受光照、温湿度、空气等影响,是否存放时限可以选取1批进行?
核查中心的回复则比较详细,具体讲到中间产品的存放时限通常应在产品上市前确定,可以在中试规模或放大生产中进行存放时限研究,并在商业化规模的工艺验证中进行确认或验证。涉及工艺、设备、贮藏条件、原辅料和包材发生变更时,需要通过风险评估来判定是否需要对存放时限进行重新研究、确认或验证。“一般而言,需采用多批代表性批次样品进行存放时限研究,也可以基于物料的特性和其他方面的风险评估来确定研究批次。”目前,主要通过稳定性试验积累数据确定存放时限,某些特殊情形下,也可通过使用前再检测的方式来确保其能够继续用于生产。”还列举了中国GMP、WHO TRS 992 Annex 4 General guidance on hold-time studies (存放时限研究通用指南)、EMA Manufacture of the finished dosage form (成品制剂的生产) 和FDA关于中间产品存放时限相关的要求。甚至还提及了可接受的最长存放时限并非指即将超出可接受标准的临界存放时限(非常重要)!
标题:存放时限验证
咨询时间:2023-08-02
咨询内容:WHO指南规定可选取1-3批进行中间产品存放时限验证,具体批次数如何评估选择?若产品工艺简单、物料性质稳定、不易受光照、温湿度、空气等影响,是否存放时限可以选取1批进行
国家药监局核查中心回复:您好。中间产品的存放时限通常应在产品上市前确定,可以在中试规模或放大生产中进行存放时限研究,并在商业化规模的工艺验证中进行确认或验证。此外,涉及工艺、设备、贮藏条件、原辅料和包材发生变更时,需要通过风险评估来判定是否需要对存放时限进行重新研究、确认或验证。一般而言,需采用多批代表性批次样品进行存放时限研究,也可以基于物料的特性和其他方面的风险评估来确定研究批次。目前,主要通过稳定性试验积累数据确定存放时限,某些特殊情形下,也可通过使用前再检测的方式来确保其能够继续用于生产。WHO TRS 992 Annex 4 General guidance on hold-time studies (存放时限研究通用指南)指出,生产过程中各物料应存放在适当的条件下,确保存放条件(包括环境和时限)对后续生产、中间产品/待包装产品和成品质量不会产生不良影响。WHO要求确定可接受的最长存放时限(注:并非指即将超出可接受标准的临界存放时限)。通常可选择1个或多个批次的物料、中间产品或待包装产品来确定存放时限,同时也要求结合物料的特性和其他相关方面,通过风险评估来确定适当的批次数。值得注意的是,WHO的指南并非要求进行中间产品存放时限验证,而是要求存放时限需经过研究后确定。世界各个国家的相关法规或指南中均对中间产品储存有相关要求,但也多未提出具体批次数。例如,FDA要求,如果生产商计划储存待包装药品,应提供稳定性数据,以证明在所述容器中延长储存时间不会对药品产生不利影响,但未规定考察的批次数。EMA Manufacture of the finished dosage form (成品制剂的生产) 指出,通常延长口服固体制剂(中间产品)储存时间超过30天、无菌产品超过24小时,如有必要,应提供至少2批试验批的存放时间稳定性研究数据。我国《药品生产质量管理规范》(2010年修订) 第二百三十二条规定,持续稳定性考察主要针对市售包装药品,但也需兼顾待包装产品。例如,当待包装产品在完成包装前,或从生产厂运输到包装厂,还需要长期贮存时,应当在相应的环境条件下,评估其对包装后产品稳定性的影响。此外,还应当考虑对贮存时间较长的中间产品进行考察。总之,中间产品存放时限的确认或验证未规定具体批次数,药品上市许可持有人应当基于对品种的理解和知识来确认选定的批次数,并使用科学的方法进行确认或验证。
WHO TRS 992 Annex 4
存放时间研究通用指南
1.概述和背景
生产商必须保证其生产的产品安全有效,具有其既定用途所需要的质量。应有一个系统来保证药品是根据经过验证的工艺和既定的程序生产的。生产工艺应具备持续生产出具有所需的质量,并符合其质量标准的药品的能力。
良好生产规范(GMP)要求必须做出适当的安排来保证分好的原料、包材、中间产品、散装产品和包装后的成品存贮在适当的条件下。存贮条件不应对随后的工艺、稳定性、安全性、有效性或起始原料、中间产品和包装前的散装产品的质量产生不良影响。因此应设定最长的可接受保存时间来保证中间产品和散装产品可以在下一工序前的保存不会使得原料质量超出可接受标准。通常,中间产品和散装产品存贮时间不应超过设定的保存时间。
最长保存时间的选择应有相应的数据来支持。研究过程可以延长以超过预定的最长时间,但没有必要将检测延长直到检验结果超出标准。
2. 术语
本指南中所用的一些重要术语定义如下。他们可能在其它语境中有不同含义。
散装产品:完成了所有工序直到,但还没有完成最终包装的制剂成品。
中间产品:完成部分工序,必须要经过进一步生产工序才能成为散装成品的部分加工产品。
3.范围
这些指南主要关注非无菌固体制剂保存时间研究设计中要考虑的问题。其中描述的许多原则也适用于其它剂型,例如液体、乳霜和软膏。这些指南不包括清洁验证或原料药或生物制剂的生产中保留时间的问题。
这些指南意在作为一个药品生产商和GMP检查员使用的基本指南。本文件无意描述建立保存时间的流程,而只是反映在保存时间研究设计中所需考虑的问题。
生产商应收集可以论证的科学数据来证明分装后的原料和包材、中间产品和散装产品:
在加工至下一工序时保持适当的质量
符合可接受标准
制剂成品应符合放行标准。
4. 需要考虑的问题
保存时间可以认为是物料(预处理后原辅料、中间产品和散装待包装的制剂)在指定条件下可以保存并能维持其质量在既定质量标准内的既定时间长度。
保存时间研究建立不同生产工序物料保存的时间限度,以保证产品的质量不会在保存时间内超出可接受标准。研究设计应反映各工序的保存时间。
保存时间通常应在产品上市前确定。工艺、设备、存贮条件、起始物料或包材变更的风险评估应包括是否需要进一步进行保存时间研究的评估。保存时间研究可以包括在研发过程中的中试规模中,也可以是在放大生产过程中,还应该包括在商业规模工艺验证中。也可以从生产过程发生的偏差调查中收集更多信息。
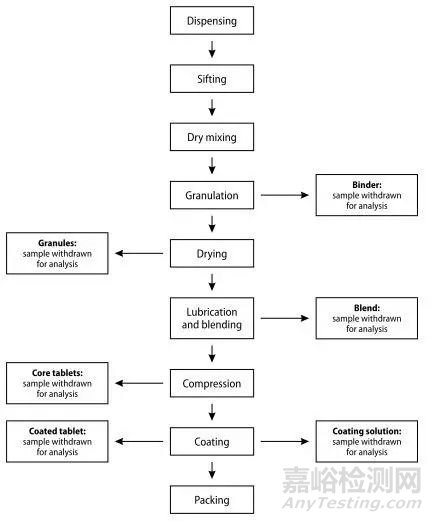
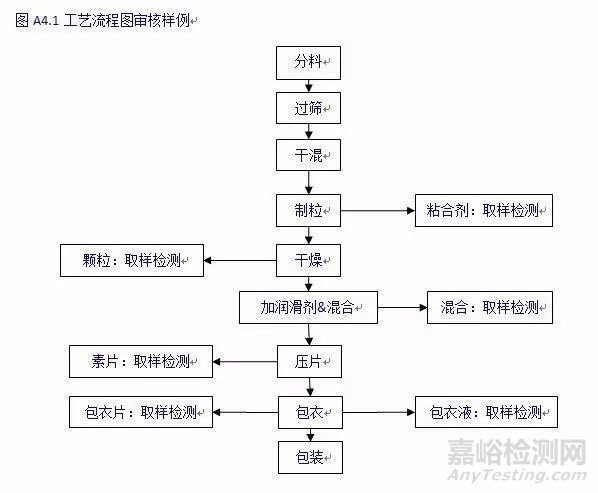
生产商可以使用工艺流程图对生产过程进行审核,将生产工艺的关键工序根据需要特殊存贮和加工过程的时间,以及环境和存贮条件的潜在影响进行划分,通常物料会在生产周期中进行停顿。
例如对于口服包衣片剂,可以考虑以下工序:
粘合剂制备到制粒---考虑颗粒
湿法制粒到干燥----考虑干燥后的颗粒
干燥后的颗粒到润滑混合---考虑有润滑的混合
混合到压片
压片到包衣----考虑片芯
包衣溶液到制备---考虑包衣溶液
包衣到包装---考虑散装包衣片
包衣到包装至散装成品
散装包装至制剂成品
Figure A4.1 Example of a flow chart for reviewing the manufacturing procedure
应遵循一份书面的方案、程序或计划,其中应包括,例如,要实施的活动、适合于物料或产品的检验项目和可接受标准。方案和报告通常应包括以下内容:标题、文件编号、版本号、日期、目标、范围、职责、流程、物料或产品的描述、样品量、取样方法和标准、可接受限度、取样频次、取样点、取样规则、存贮条件、容器类型、分析方法、结果、结论、建议、签名和日期。可接受标准通常会比注册的质量标准更严格,以保证原料很好地受到控制。在设定质量标准时,需要考虑所有已知的稳定性趋势。
对于特定的产品,还需要考虑微生物方面的问题,适当时应包括在研究中。
所有散装中间产品和产品的检测应使用经过验证的可以指示稳定性的方法。
通常可以使用一批或多批物料、中间产品或产品来确定保存时间。可以使用基于风险的方法来确定批次数,同时考虑物料的特性和其它相关方面。进行保存时间研究的物料或产品批次的代表性样品要保存至指定的时长。每类物料的保存时间应根据保存在生产中使用的原装或模拟容器中的物料的研究制订。用于保存时间研究的样品存贮所用的容器应与生产中所用的包装相同,除非该包装实在太大,这时应使用一个相等同的容器(材质相同,使用与生产相同的密闭系统)。如果在保存时间测试中必须减小容器尺寸,则应进行论证。
生产和/或暂存过程中用于散装存贮的容器的顶部空间非常重要,例如,因为氧化引起可能的降解风险,则保存时间研究应代表最差情形时的条件。在这种情况下,顶部空间与受试容器容量的比例应至少大于常规生产中可能的最大情形(特别要考虑半满容器)。样品存贮的环境条件应与暂存区域/生产工序的条件相同。应建立并遵守取样计划,以在不同时间间隔取样检测。应根据批量、时间间隔和要检验的项目计算所需的样品数量。应装结果与受控样品的初始数据进行比较。适当时,样品可以放在一起分析,例如,将样品合并分析不会导致单独样品分析可检出的问题被忽略时。
适当时,应将所获得的数据进行统计学研究,以找出趋势,判定限度和保存时间对应关系。
从中间产品或散装产品生产,并经过保存时间研究的制剂批次可以考虑用作长期稳定性试验,以查看在中间时间点到货架期结束时的数据是否显示出不良趋势。产品的货架时间---不考虑保存时间---应从活性成分与其它成分混合的时间开始计算。通常,中间产品和散装产品的存贮时间不应超过所建立的保存时间。
表A4.1给出了在包衣片中可以考虑的工序、研究时间和检测的例子。
表A4.1: 根据风险评估和特定的产品需求拟定的工序、研究时间点举例
|
工序
|
根据质量标准要检测的项目
|
研究时间点
|
|
粘合剂制备
|
微生物测试、外观、粘度,适用时
|
第0、2、5、8小时
淀粉:第0、2、5小时
|
|
分散剂制备(包括制粒糊、包衣溶液和包衣混悬液)
|
物理外观、比重、粘度、沉淀、微生物测试
|
第0、12、24、36、48、60、72小时
|
|
颗粒
|
性状、含量、降解产品/相关物质、干燥失策重、水分、粒径分布、堆密度、击拍密度、休止角
|
第0、15、30、45天
|
|
混合
|
微生物测试、干燥失重、混合均一性、粒径分布、堆密度/击拍密度
|
第0、15、30、45天
|
|
素片---未包衣(在散装容器中)
|
性状、硬度、厚度、脆碎度、分散度、溶出度或溶出概况、含量、降解产品/相关物质、剂量均匀性、微生物测试
|
第0、30、45、60、90天
|
|
包衣片(散装容器内)
|
性状、外观或目视检查、硬度、厚度、脆碎度、分散度、溶出度或溶出概况、含量、降解产品/相关物质、水分、微生物测试
|
第0、30、45、60、90天
|
参考文献
GMP补充指南:验证,非无菌工艺验证。WHO专家委员会制剂质量标准:第49号报告,日内瓦,WHO,2015:附录3(WHO技术报告第992号)。