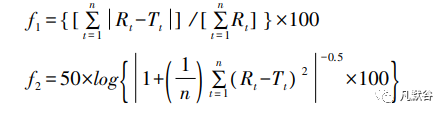摘要
随着中药制剂现代化的发展,溶出度作为固体制剂体外工艺控制的重要指标,其日益受到重视。针对中药药效成分的复杂多样性,本文依据文献对中药固体制剂溶出度测定的重要意义、溶出度测定装置、检测方法和数据处理分析方法的现状进行综述。提出用不同批次制剂的溶出曲线点的宏定性相似度Sm≥0.9 和宏定量相似度 70%≤Pm≤110%作为判定批间溶出曲线一致性的新标准,为中药固体制剂的溶出度研究提出的控制标准。评价中药工艺是否恒定规范以保证药效是否最优,固体制剂溶出度的一致性是先决条件。中药一致性控制中的药效物质总量控制并不难,最难的地方是中药固体溶出度的一致性控制。溶出度控制是中药固体制剂一致性控制的重点和难点,是制剂药效一致性控制的前提条件。
关键词
中药一致性评价; 中药固体制剂;溶出度测定方法; 紫外全指纹溶出度测定法;药效物质总量一致性; 固体制剂溶出度一致性
溶出度是指在规定条件下,药效活性物质从普通固体制剂中溶出的速度和程度,对于缓、控释制剂等则称为释放度,作为控制固体制剂工艺一致性的体外评价方法,1970 年《美国药典》( 18 版) 首次收载泼尼松片等品种的溶出度检测,《中 国 药 典》( 1977 年版) 开始收载溶出度检测项目[1]。2015 年国家食品药品监督管理总局发布的《普通口服固体制剂溶出度试验技术指导原则》进一步规范了溶出度测定方法。中药固体剂型包括散剂、丸剂、胶囊剂和片剂等,近年来随着新辅料和新技术等的应用,出现新型固体制剂如缓( 控) 释制剂、择时和靶向释药系统等。药品作为特殊商品,其质量是确保用药安全和有效的关键,而中药制剂存在药材来源分散,加工炮制方法和制剂生产工艺不统一等因素,造成其质量和药效的不确定性,因此,中药一致性评价的溶出度问题正成为确保药品药效均一稳定的关键技术问题。2016 年 3 月 5 日国务院发布《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见( 国办发〔2016〕8 号) 》指出,开展仿制药质量和疗效一致性评价工作,对提升我国制药行业整体水平,保障药品安全性和有效性,促进医药产业升级和结构调整,增强国际竞争能力,都具有十分重要的意义”。溶出度作为评价固体制剂内在质量的关键指标显得尤为重要,由于与化学药的药效成分明确不同,中药制剂的药效物质复杂多样,如何建立合适的溶出度研究方法成为当前中药固体制剂制备、质量控制和一致性评价的热点之一[2-4]。本文通过文献查阅,对溶出度测定意义、溶出度测定装置、检测方法和数据处理分析方法进行综述,为中药固体制剂的溶出度研究提供新参考和新思路
1、溶出度研究的意义
1.1 溶出度是药品质量控制的重要指标
固体制剂存在生物利用度低、差异性大等问题,造成同一品种制剂生物不等效,美国食品药品监督管理局记载的生物不等效药品中,约 80%存在溶出问题[5]。为确保同一品种药品间、批次间的质量和疗效一致,采用溶出度试验进行药品质量一致性评价成为研究的重要 内 容[6-11]。闫 慧 等[7] 采用高效液相色谱( HPLC) 指纹图谱和紫外全指纹溶出度测定法对不同厂家复方甘草片的物质组成并对溶出度进行测定,以最后取样点的溶出液紫外图谱作为制剂全溶出标准图谱,将不同时间点溶出样品的紫外图谱经“复方甘草片主组分一致性数字化评价系统 1.0”软件,以 210~400 nm 的 211 个波长下的紫外吸收光度点来计算溶出度和评价溶出曲线的一致性,发现各厂家制剂的 28 个 HPLC 主组分指纹的宏定量相似度很接近即药效物质总量都控制得很好,但其溶出行为存在显著差异,可能与甘草浸膏粉质量和生产工艺不同有直接关系。提示应从原料和制备工艺入手开展研究才能保证该制剂质量的一致性,即在复方甘草片质量一致性评价中,工艺研究和溶出度控制更为重要。
1.2 溶出度是中药剂型选择、处方设计和制备工艺优化的核心指标
1.2.1 剂型选择
中药固体制剂的剂型包括散剂、颗粒剂、片剂和丸剂等,可满足不同病症缓急的用药需要,如丸剂就有“水丸取其易化,蜜丸取其缓化,糊丸取其迟化,蜡丸取其难化”的传统用药选择规范。现代研究发现,不同剂型满足疾病缓急用药需要的根本与药物溶出行为相关,溶出度测定可为剂型选择提供依据。叶英响等[12]采用桨法,测定不同类型六味地黄丸( 分别为水泛丸、浓缩丸、蜜丸、水蜜丸、糊丸和蜡丸) 的溶出度,发现不同类型丸剂的溶出行为与丸剂传统用药选择规范一致。剂型选择基本代表药物的作用吸收部位,要根据体内吸收部位确定溶出介质的种类和 pH,也是做好溶出方法开发的前提。
1.2.2 处方设计
新辅料的开发应用促进了中药制剂现代化,辅料对制剂质量如溶出行为影响显著。以溶出度试验指导开发新辅料、制剂辅料的选择和处 方 设 计,都可有效提升中药固 体制剂的质量[13-14]。蔡延渠等[15]对桃胶进行改良,制备喘平方缓释片,采用小杯法,以麻黄碱、伪麻黄碱和东莨菪碱为测定指标,对其释药曲线进行研究,发现各成分可实现同步释放,改良桃胶缓释性能更显著,这与改良桃胶的含糖量增加,水溶性和吸水性提高,溶胀性能和形成水凝胶强度提高有关,可作为新型缓释辅料应用。
1.2.3 制备工艺
随着“药物一致性评价”研究的深入开展,制备工艺对制剂溶出行为的影响日益受到关注,探讨制剂技术和工艺对溶出度的影响,对合理选择制备工艺和规范工艺参数具有指导意义[11,16-19]。张嫱等[11]采用桨法,在模拟消化道体液的 4 种介质( 分别为 pH 1.2、pH 4.0、pH 6.8 缓冲液和纯水) 中,对不同厂家,不同批号复方丹参片的丹参酮ⅡA、丹酚酸 B、三七皂苷 R1、人参皂苷 Rg1等 7种成分的溶出曲线进行测定,发现制剂的溶出行为存在较大差异,可能与辅料组成和制粒等工艺步骤控制不同有直接关系,提示中药固体制剂的工艺条件选择应结合其溶出度测定数据来最终确定合理工艺参数。
1.3 溶出度可作为预测体内生物利用度的重要指标
生物利用度是指制剂中药物吸收进入体循环的速度和程度,与其药效和安全性直接相关,需通过体内试验进行评价,存在耗时、成本高等问题[20]。中药固体制剂的药效成分只有在胃内充分溶出后,才能吸收进入血液发挥药效,通过制剂体外溶出与体内吸收的相关性研究,可由简捷的溶出度测定来预测其生物利用度[21-23]。赖宏强等[24]对川芎组分片的体外溶出样品和大鼠体内血浆样品进行紫外扫描( 波长范围为 200 ~ 600 nm) ,根据吸光度对波长积分得到曲线下面积( 吸波面积) ,计算制剂的体外累积溶出度和体内累积吸收百分率,两组数据相关性良好,实现由体外溶出来预测体内生物利用度。但该法要排除体内内源物质所产生的紫外吸收才妥。
2、中药溶出度测定装置
《中国药典》2015 年版收载溶出度测定装置有篮法、桨法、小杯法、桨碟法和转筒法,2020 年版新增流池法。篮法和桨法的装置简单、耐用,易实现自动化检测,应用广泛[9-10,12,25]; 对于微粒给药系统如纳米粒、脂质体等,需将制剂置于透析袋中进行测定[26]; 溶出介质由水相和有机相组成,则称双相法,用于水难溶性药物的测定[5]。小杯法的介质用量少,可用于剂量低的制剂[27-28]。桨碟法和转筒法可用于透皮贴剂[29-30]; 流池法可通过调节溶出介质的流动方式有效模拟体内环境,有开放式和循环式两种[31]。溶出杯本身就是一个仿生试验装置,900 mL溶出介质的体积基本模仿人胃的体积,37 ℃ 也是人的正常体温范围内,溶出仪的旋转浆模仿人胃的蠕动。
考虑到固体制剂进入体内胃肠道后的溶出和吸收过程具有连续、动态、同步的特征,近年有学者提出设计药物溶出/吸收仿生评价系统,模拟人体胃肠道变化条件下制剂的药物溶出和跨膜吸收过程,实现对制剂中药物连续、动态释放和跨膜转运性能的研究,与传统溶出度测定装置相比,更接近体内实际环境[32-34]。
3、 中药溶出度测定方法
3.1 检测指标
3.1.1 单指标成分
选择制剂中单个或一类成分 ( 如总黄酮、总生物碱和总皂苷等) 作为检测指标, 适用于药效成分明确或单一成分制备的中药制 剂[8,26,28]。刘芳芳等[25]以总黄酮苷为指标制定了 创新中药奥兰替胃康片的体外溶出度测定方法。复 方制剂也可用主要药效成分的单指标作为测定溶出 度的控制指标。这种方法属于抓主要矛盾的方法, 但评价结果带有一定的片面性。
3.1.2 多指标成分
选择制剂中多个或多类成分 作为检测指标,与单指标相比,更能体现中药多成分 的特色,并且对不同指标成分的溶出曲线进行比较, 可评价中药制剂的多成分均衡释药性能,但多指标 成分测定存在指标筛选、测定时间长如 HPLC 法需 梯度洗脱、指标成分的溶出特性不能完全代表中药 全成分等问题[35-36]。段晓颖等[29]采用桨碟法,通 过相似因子 f2法对生物黏附制剂愈溃膜中丹皮酚、 靛玉红和龙脑 3 个指标的溶出行为进行评价,发现 制剂具有均衡释药特性。监控中药多指标成分的溶 出度采用 HPLC 指纹图谱是很好方法,多指标成分 间或与相邻峰首先要满足分离度都大于 2。显然多 指标成分的溶出度需要建立各指标的标准曲线法, 闫慧[37]提出用一线多评法可节省对照品,其准确性 可控且好于一测多评法。
3.1.3 中药物质组
中药物质组是将基因组学、蛋白质组学等系统生物学技术与中医药整体观结合提出的新概念,采用中药物质组作为检测指标,对制剂的溶出行为进行评价,与单一或少数指标成分相比,更能体现中药多组成和整体性的特点,适用于多组分中药固体制剂的溶出度研究[12,38-41]。凌昳等[41]采用紫外吸收光谱法对银翘解毒丸的溶出样品进行测定,经 Kalman 滤波法处理,定量描述整体和不同程度切割后丸剂的物质组溶出度,并以取样间隔物质组的归一化溶出增量为指标,对制剂的物质组药物溶出同步性进行比较,发现采用物质组为指标可定量描述银翘解毒大蜜丸中药物的溶出特性和同步性,物质组的溶出速度和同步性与丸剂切割程度直接相关,过分切割影响制剂中物质组的缓慢溶出特性和同步性。中药物质组的溶出度可采用 HPLC 指纹图谱法来一次实现测定物质组的整体和各单一组分的溶出度。中药物质组的各单一组分的溶出曲线可能存在差异,可选择合适辅料和制剂工艺加以调控。
3.1.4 中药全成分
基于中药制剂成分的复杂多样性,其质量控制时应体现“整体观”思路,测定单一或数个指标成分溶出行为不能充分体现制剂药效成分 整 体 的 溶 出 特 性[42-43]。近年来出现了采用HPLC 指纹图谱[44-46]、生物活性检测[47-50]、紫外全指纹溶出度法[7,51-52]等方法,进行中药制剂全成分的溶出检测。孙国祥课题组[7,51-52]开发的 HPLC 指纹图谱整体检测溶出度法和紫外全指纹溶出度测定法,具有操作简便、快速、适用范围广等优势,测定的关键步骤是制剂全溶出液的制备,可采用制剂溶出度测定 120 min 时的溶出液作为全溶出溶液,考虑到有些制剂的中药整体组分不一定完全溶出,以此溶出液指纹图谱测定作为基准只能得到相对溶出度数据。采用将 10 片固体制剂投入到 900 mL 的 37℃溶出介质中超声 20 ~ 30 min,然后取样,稀释 10倍后,测定所得图谱作为制剂全溶出液的标准图谱,进行溶出度测定和计算。溶出度检查时,可测定相应时间溶出液的指纹图谱和全溶出液指纹图谱的宏定量相似度作为制剂的溶出度的数据。缺少 DAD检测器时可采用一个能兼顾多个组分吸收的最大吸收波长来同法测定。
3.2 检测技术
3.2.1 气相色谱法
气相色谱法是利用气体流动相和色谱柱实现组分分离的分析方法,高效快捷,可用于易挥发性中药成分的检测[29,53-55]。王振等[55]将冰片包合于 β-环糊精中制得复方丹参肠黏附微丸,采用篮法、气相色谱法测定制剂在模拟胃、肠液中的龙脑溶出度,与复方丹参片相比,丸剂 12 h 累积释放近 70%,具有缓释特性。
3.2.2 紫外分光光度法
紫外分光光度法是基于溶液中药物成分对紫外线或可见光的吸收程度与其浓度之间定量关系的分析方法,具有操作简单、分析速度快等优势,可用于具有紫外吸收的单一成分或物质组的检测。王丽琼等[56] 以 250 nm 为检测波长,采用紫外法测定,经加含量值校正的自身对照法计算溶出数据,与 HPLC 对照品法相比,两者无显著性差异。
针对中药制剂成分复杂,溶出液紫外光谱多为混合光谱,专属性较差,但可反映制剂整体成分的溶出行为,近年来,有学者提出了紫外指纹图谱的新技术,用于制剂中整体成分或物质组的溶出度测定[7,12,40,51-52]。叶英响等[12]以物质组为检测指标,对不同类型六味地黄丸的溶出样品进行紫外扫描,以物质组浓度标准谱为基准,计算其释放度,并绘制物质组释放度二维图谱和物质组释放增量三维图谱,实现对丸剂物质组溶出行为的可视和定量描述,发现不同丸剂的溶出行为存在差异性和相似性。
3.2.3 高效液相色谱法
高效液相色谱法作为中药制剂质量检测常用方法,专属性强,灵敏度高,可用于单一成分[8,10,28]、多成分[57-60]和指纹图谱全成分[44-46]的测定,其中指纹图谱通过整体化学指纹的溶出动力学评价制剂质量,体现了中药整体性的特色[61]。杨岩涛等[45]采用 HPLC 指纹图谱法测定补阳还五单室渗透泵缓释片的体外释放度,将图谱数据按出峰时间 10 min 为间隔分段,按总量统计矩原理计算并评价成分间释药相似性,发现缓释片可实现“整体受控、同步释放”,同时探讨超分子印迹模板在中药不同成分整体同步释放中的应用。
3.2.4 生物效应法
中药制剂药效通常是多组分协同作用的结果,反映其药效作用的整体性,有学者提出采用生物效应法代替单个( 类) 或几个( 类) 指标成分的检测,从生物活性角度客观评价制剂的溶出行为,生物效应检测方法的选择需遵循“相关性、重复性、灵敏性和适用性”的原则,此方法与制剂药效作 用 相 一 致,但存在操作烦琐和适用性等问题[47-50]。马晓斐等[62]进行复方丹参片溶出度研究时,经实时细胞电子分析技术( RTCA) 实验和 CCK-8 实验联合筛选得到对制剂具有特定依赖性的H9C2 细胞作为检测用细胞,并确定作用于细胞的合适药效成分浓度范围; 将不同时间的溶出样品作用于细胞,采用 RTCA 监测细胞的动态变化,转换成电信号细胞指数( CI) ,得到时间剂量依赖性细胞反应曲线,通过 CI 值计算制剂的累积溶出率,进行数学模型拟合,与紫外分光光度法进行比较,发现两种方法具有一定的相关性,溶出数据符合 Weibull 模型,RTCA 法的模型拟合相关性优于紫外法。生物效应法在各个方面应用都只是一个美好的理想,真正用于生产检验的难度是很大的。任何生物效应法都只是中药总效应的一个侧面,是一种单一活性方向的生物效应。
3.2.5 其他技术
随着检测技术、新制剂的发展和制剂溶出规律及控制体系研究的需要,新技术不断用于固体制剂溶出度的研究。梁兰等[63]在黄柏煎煮提取过程,采用光谱成像技术测定在不同煎煮时间黄柏样品的光谱图像,根据荧光强度评价黄柏煎煮过程中活性成分的扩散溶出行为,该技术可推广应用于缓、控释制剂的释药行为、机理和控制体系的研究。近红外透射光谱法[64]和拉曼光谱法[65]已用于化学药制剂的溶出度研究。Ahme 等[66]采用电位分析法监测制剂中伪麻黄碱和布洛芬的溶出行为,此方法操作简便,但易受其他成分干扰,多用于成分简单的制剂测定,如测定成分复杂的中药制剂,应结合化学计算学方法以消除干扰。
紫外成像技术可采集制剂如片剂、凝胶剂等溶出过程的图像,观察制剂表层溶解和溶蚀情况,结合药物溶出数据,探讨制剂释药机制[67]。采用同步辐射显微 CT 可连续观察释药过程中固体制剂的形态结构变化,探讨制剂结构因素对药物释放的影响和释药机制[68]。固体制剂的释药机制和溶出影响因素的研究,可指导新剂型的设计。
4、中药溶出度数据处理分析方法
中药固体制剂溶出度测定相关数据的处理分析,可用于评价药品质量一致性、多成分释药均衡性和药物释药机制的探讨等。
4.1 差异因子( f1 ) 和相似因子( f2 ) 法
作为非模型依赖法,通过计算 f1值和 f2值,可评价不同药物的溶出行为一致性,也可用于中药制剂不同成分间溶出同 步 性 的 比 较,评价制剂中多成分释药的均衡性[8-9,27]。
其中 n 为取样时间点个数,Rt和 Tt分别为参比和试验样品在 t 时间的溶出数据。差异因子( f1 ) 是计算两条溶出曲线在每一时间点的差异,来衡量两条曲线相对偏差的参数。f1实际上是溶出曲线上每个点的溶出度的相对平均偏差,是一个误差值。相似因子( f2 ) 是衡量两条溶出曲线相似度的参数,通常 f1值小于 15 或 f2值大于 50,则认为两条溶出曲线具有相似性。相似因子( f2 ) 法对差异限度要求过高,监控溶出曲线一致性差别的效果特别明显,易显示溶出曲线的差异性。相似因子( f2 ) 法对于多组分的中药有很多不适应性,就是中药用相似因子( f2 )评价可能要求过高、过严格,按照中药的特征性需要使用程度溶出度法。
4.2 溶出模型拟合法
采用不同数学模型( 包括零级方 程、一 级 方 程、Weibull 方 程、Higuchi 方 程、Ritger-peppas 方程等) 对制剂溶出数据进行拟合,通过拟合优度参数比较确定最优模型,根据最优模型参数如 Weibull 方程中 T50和 Td( 分别为制剂中药物溶出 50%和 63.2%的时间) 等对不同溶出曲线的相似性进行比较,还可根据 模型参数如 Ritger -peppas 模型中释放参数 n 值大小推测制剂的药物释放机制[12,62,69-70]。
其中Mt /M!为制剂 t 时间的累积溶出百分率,k为常数,n 为释放参数。
4.3 Kalman 滤波法
Kalman 滤波法是基于经典的最优化递推估算法[71],当中药制剂中复杂药效成分可视为同一物质组,并且各组分可实现同步释放时,与紫外分光光度法联用,对制剂中物质组进行定量分析[12,40-41,69,72]。吴素香等[40]测定桂枝茯苓丸的溶出行为,分别采用 HPLC 法测定肉桂酸、芍药苷、丹皮酚 3 种有效成分和 Kalman 滤波-UV 法测定物质组作为溶出检测指标,发现丸剂有效成分与物质组的溶出相关性良好,物质组可代表制剂整体成分描述丸剂的释药特征。
4.4 中药溶出度的定量指纹测定法
指纹图谱用于中药制剂溶出度研究时,通常采用指纹图谱曲线下面积表示一定物质量下的响应总量,按谱量学方法对总成分进行定量分析,计算制剂中每个指纹成分和整体成分的溶出度,常用指纹图谱有 HPLC 指纹图谱和 UV 全波长指纹图谱等[51-52,61,73]。孙林艳等[52]以 小 杯 法,采用流动注射法 ( 流 速 为 0. 5mL·min-1) 对 20 批复方两面针含片的不同时间溶出液进行紫外扫描( 波长 190 ~ 400 nm) ,以最后取样时间的图谱为基准,采用“中药溶出紫外指纹图谱定量相似度数字化评价系统 3.0”软件计算制剂溶出度,45 min 时溶出度均超过 70%。
孙国祥课题组[7,74]对全国 14 家以上厂家的复方甘草片进行了紫外全指纹溶出度测定,基于标准制剂结合定量指纹图谱和多 Q-markers 的精准定量控制,提出在相同溶出试验条件下,用不同批次固体制剂的全部溶出液的宏定性相似度 Sm≥0.9 和宏定量相似度 70%≤Pm≤110%作为判定中药溶出曲线的一致性的新方法和评价新标准,该方法收录在“中药主组分一致性数字化评价系统 3.0”软件中,考虑到大多数中药成分具有紫外吸收的特性,监控190~400 nm 的全紫外吸收成分的溶出情况,可体现中药整体成分的溶出状态,并且操作简便快捷,测定1 个样品的时间约 1 min,为大量溶出数据的测定节约了宝贵时间,并可计算溶出曲线相似性因子 f2,适用于化学药和中药固体制剂一致性评价中溶出度曲线相似性的计算。对于取液后的“补液法”和“弃液法”可对应采取不同的计算公式和校正处理方法。
在复方甘草片质量一致性评价中,选择北京国药工业集团有限公司的 3 批复方甘草片作为对照制剂,以水为溶出介质,评价广州粤华制药有限公司的3 批申报制剂的溶出曲线的宏定性相似度均 Sm≥0.90和宏定量相似度均 70%≤Pm ≤110%,分别为81.0%、75.8%和83.8%,据此,我们判定广州粤华的复方甘草片在水中溶出曲线与北京国药的溶出曲线相似,这个方法的好处是把被评价厂家的溶出情况给出一个程度溶出度 Pm,它能详细底揭示被评制剂的具体溶出情况( 溶出药效物质总量情况) 。
4.5 组分权重系数法
根据中药多成分的特点,有学者提出将制剂中各成分效应( 如物质量、药效等)与总效应之间的比值作为权重系数,对各成分的溶出曲线进行加权得到整合溶出曲线,表征制剂整体的溶出行为[23,75-76]。郑娟等[75]采用 HPLC 法测定六味五灵片中五味子甲素、特女贞苷和连翘苷的溶出度,并根据各成分的质量权重得到制剂的整合溶出度,同时基于制剂溶出液对 LX-2 肝星状细胞的抑制率得到制剂的生物效应溶出度,采用相似因子法对各单一成分和整合溶出度与生物效应溶出度进行比较,发现整合溶出度与生物效应溶出度相似性最好,可反映制剂的整体溶出特性。
5、结论
中药固体制剂一致性评价的关键问题之一是溶出度的控制与测定,采用的方法应能评价中药复杂样品体系整体组分溶出的一致性,能够考量制剂工艺的重复性和恒定性。中药固体制剂在完成药效总物质的恒定性和一致性控制后,关键问题应该是溶出度一致性控制的问题,因为溶出度直接关系到药效的重现性和稳定性。中药制剂药效物质总量的一致性固然重要,但控制并不难,最难的应是溶出度的一致性控制,药效物质的溶出一致性才是保证药效一致性的必要基础。中药一致性评价的重点和难点应是溶出度控制,它是比总药效物质控制更难的一个技术问题,也是更重要的关键一步。
溶出度作为固体制剂体外质量评价的重要指标应用日益广泛,针对中药药效成分复杂多样的特点,在溶出度研究过程中应注重整体性,物质组和全成分指纹图谱法、生物效应法等技术的应用可实现中药制剂整体溶出特性评价的要求,可根据药效物质特性选择相应的溶出检测方法,同时有些问题还需进一步探讨,如指纹图谱法依据指纹峰进行溶出度评价,如何确立指纹峰? 指纹峰对应物质是否与药效作用相关等? 需与中药药效物质基础研究相结合。多学科交叉融合,如色谱技术、光谱成像技术、化学计量学、机械设备技术等,可提升中药固体制剂溶出度的研究水平和应用价值,实现制剂溶出度快速在线检测,探讨制剂药效成分的溶出机制和控制因素,保证现代中药固体制剂的质量一致性和可控性。本文推荐使用紫外全指纹溶出度测定法,该法在我国第一个主要成分为中药组分的复方甘草片质量一致性评价中获得成功应用。